
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дефект транспорта карнитина - причины, диагностика, лечение
Дефект транспорта карнитина - первичная недостаточность карнитина обусловлена генетическим нарушением переносчика данной аминокислоты. У новорожденных и детей 1-го года жизни болезнь проявляется в виде гипогликемии, непереносимости голодания, в ряде случаев может приводить к внезапной смерти.
Мышечная слабость и прогресссирующая кардиомиопатия характерны для пациентов более старшего возраста. В плазме крови концентрация карнитина снижается до уровня менее 10% нормативных значений. Установление диагноза основывается на нагрузочных тестах в культуре кожных фибробластов и/или молекулярно-генетическом анализе гена OCTN2.
Дефицит карнитин-пальмитоилтрансферазы-1 (СРТ-1). Болезнь описана в 1981 году. Выражается в виде некетотической гипогликемии, провоцируемой голоданием; сопровождается наличием в крови высокой концентрации свободного и связанного карнитина. Характерные метаболиты ЖК в крови и моче отсутствуют, что является важным диагностическим признаком заболевания. Описано свыше 30 случаев с преимущественным поражением печени, но встречаются и миопатические формы заболевания.
В частности, при детской форме дефицита карнитин-пальмитоилтрансферазы-1 отмечаются сочетанное поражение печени и нарушения со стороны скелетных мышц, что отличает ее от взрослой формы (с изолированной прогрессирующей миопатией).
Дефицит карнитин-палъмитоилтрансферазы-2 (СРТП). В классическом варианте при этом виде патологии наблюдается миопатия, провоцируемая физической нагрузкой, а также миоглобинурия. У детей с миопатической формой болезни обнаруживается частая мутация 439С>Т, S113L. У гомозиготных по этой мутации пациентов остаточная активность фермента составляет около 15-25% нормальных значений, что является адекватным для нормального функционирования сердечной мышцы и печени, но недостаточным для скелетных мышц (особенно при голодании или повышенной физической активности).
При манифестации болезни в неонатальном периоде характерно полиорганное поражение и наличие врожденных аномалий, нередко приводящие к летальному исходу до окончания 1-го месяца жизни. Описана также промежуточная форма недостаточности СРТН, встречающаяся у «компаунд»-гетерозигот с мутациями средней и тяжелой степенями выраженности.
Дефицит карнитин/ацилкарнитинтранслоказы. Болезнь описана в 1992 году. Клинические проявления заболевания напоминают симптомы инфантильной формы недостаточности СРТП. Среди симптомов доминирует скелетная миопатия, приступы гипогликемии встречаются относительно редко, что в определенной степени затрудняет диагностику.
Большинство детей с дефицитом карнитин/ацилкарнитинтранслоказы впоследствии погибают вследствие хронической прогрессирующей печеночной недостаточности с гипераммониемией, а также в результате гипертрофической кардиомиопатии и септальных пороков сердца.
A.A.M. Morris и соавт. описали несколько клинических случаев с более мягкой формой болезни, чувствительной к терапии. В гене САСТ описано немало мутаций, преимущественно затрагивающих «сплайсинг»-сайты, но четкие гено-фенотипические корреляции не были установлены. Предполагается, что тандемная масс-спектроскопия позволит выявлять большинство случаев этого заболевания в ходе проведения неонатального скрининга.
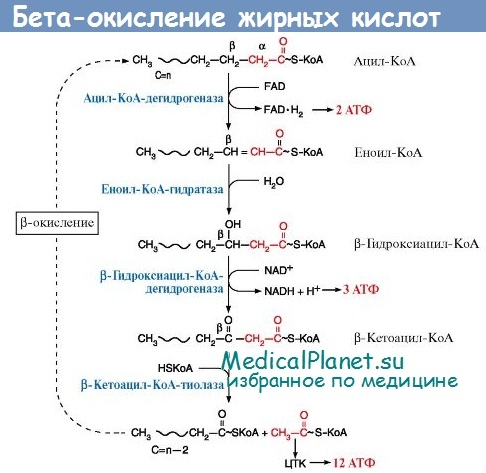
- Синдром (болезнь) Лея у новорожденных - причины, диагностика, лечение
- Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) у новорожденных - причины, диагностика, лечение
- Дефицит короткоцепочечной ацил-КоА-дегидрогеназы (short-chain acyl-CoA dehydrogenase deficiency, SCADD) - причины, диагностика, лечение
- Дефект транспорта карнитина - причины, диагностика, лечение
- Дефицит среднецепочечной ацил-КоА-дегидрогеназы (medium-chain acyl-CoA dehydrogenase deficiency, MCADD) - причины, диагностика, лечение
- Дефицит длинноцепочечной 3-гидрокси-ацил-КоА-дегидрогеназы (long-chain 3-hydroxyacyl-СоА dehydrogenase deficiency, LCHADD) - причины, диагностика, лечение
- Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - причины, диагностика, лечение
- Недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (HMG-CoA SD) - причины, диагностика, лечение
- Мукополисахаридозы (МПС) у новорожденных - причины, диагностика, лечение
- Синдром Ангельмана - причины, диагностика, лечение