
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - причины, диагностика, лечение
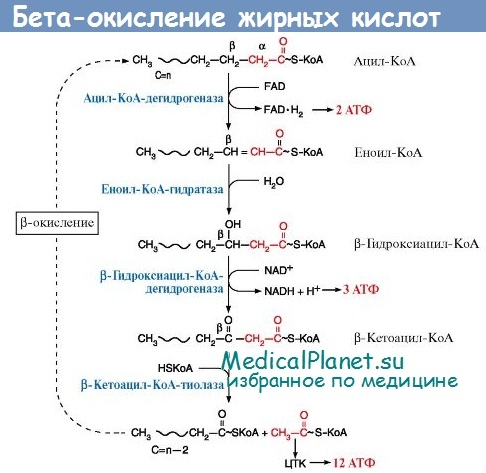
Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - представляет собой группу различных заболеваний, объединяемых по принципу наличия дефекта вхождения электронов в цепь их транспорта.
Наиболее тяжелый вариант MADD, обозначаемый MAD:S (S — от англ. severe), также носит название «глутаровая ацидурия типа II» (glutaric aciduria type II). В неонатальном периоде болезнь может закончиться летальным исходом вследствие развития глубокого ацидоза, гипогликемии, комы и полиорганных поражений. У части пациентов наличествует лицевой дисморфизм и другие признаки.
В менее тяжелых случаях MADD не отмечается дисморфизма, но отсутствует прибавка в массе тела, наличествуют мышечная гипотония, кардиомиопатия, поражение печени. Из органических кислот характерно накопление в моче этилмалоновой, глутаровой, 2-гидроксиглутаровой, адипиновой, пробковой и себациновой кислот. При скрининге аминокислот в моче отмечается увеличение содержания изовалерила, изобутирил-глицина, 2-метил-бутирил-глицина и саркозина.
Более легкий вариант болезни, получивший название MAD:M (М — от. англ. mild), по-другому называется этилмалоновая-адипиновая ацидурия (ethylmalonic-adipinic aciduria). При ней у детей появляются симптомы, типичные для других форм недостаточности ацил-КоА-дегидрогеназ, например MCADD. При этом в моче из органических кислот обычно обнаруживаются только этилмалоновая, адипиновая и метилянтарная кислоты.
В повышенном количестве встречаются дикарбоксильные кислоты, снижение которых возможно при использовании витамина В2 (отсюда название - рибофлавин-чувствительная дикарбоксилъная ацидурия, англ. riboflavin-responsive dicarboxylic aciduria). Схожий метаболический паттерн отмечается при SCADD, что затрудняет диагностику.
Существует два основных дефекта синтеза кетоновых тел: недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (3-hydroxy-3-methylglutaryl-CoA synthase deficiency, HMGCoA SD) и дефицит З-гидрокси-3-метилглутарил-КоА-лиазы (3-hydroxy-3-methylglutaryl-CoA lyase deficiency, HMG-CoA LD). Их сиптоматика практически идентична.
- Читать "Недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (HMG-CoA SD) - причины, диагностика, лечение"
Оглавление темы "Детская неврология":- Синдром (болезнь) Лея у новорожденных - причины, диагностика, лечение
- Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) у новорожденных - причины, диагностика, лечение
- Дефицит короткоцепочечной ацил-КоА-дегидрогеназы (short-chain acyl-CoA dehydrogenase deficiency, SCADD) - причины, диагностика, лечение
- Дефект транспорта карнитина - причины, диагностика, лечение
- Дефицит среднецепочечной ацил-КоА-дегидрогеназы (medium-chain acyl-CoA dehydrogenase deficiency, MCADD) - причины, диагностика, лечение
- Дефицит длинноцепочечной 3-гидрокси-ацил-КоА-дегидрогеназы (long-chain 3-hydroxyacyl-СоА dehydrogenase deficiency, LCHADD) - причины, диагностика, лечение
- Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - причины, диагностика, лечение
- Недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (HMG-CoA SD) - причины, диагностика, лечение
- Мукополисахаридозы (МПС) у новорожденных - причины, диагностика, лечение
- Синдром Ангельмана - причины, диагностика, лечение