
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Мукополисахаридозы (МПС) у новорожденных - причины, диагностика, лечение
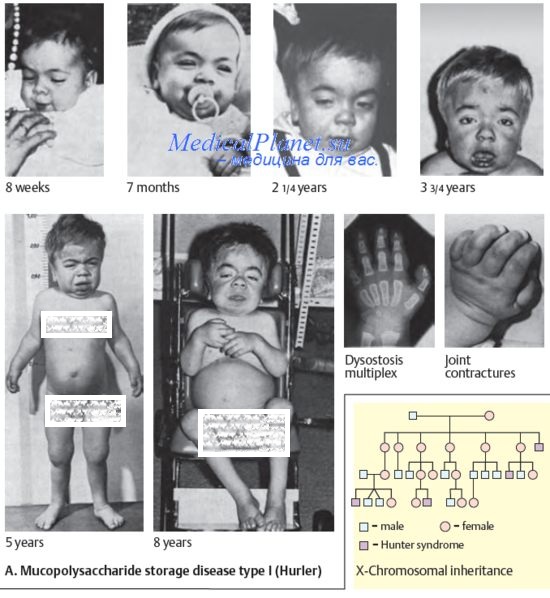
Мукополисахаридозы — группа метаболических заболеваний, вызываемых отсутствием или нарушением функций лизосомных ферментов, необходимых для расщепления гликозаминогликанов (мукополисахаридов). МПС (англ. mucopolysaccharidoses) относятся к группе лизосомных (лизосомальных) генетических болезней, которых в настоящее время насчитывается свыше 40.
Всего к настоящему времени известны 7 основных разновидностей мукополисахаридозов: тип I (синдром Гурлер — тип I Н, синдром Гурлер-Шиайа — тип I HS, синдром Шиайа — тип I S), тип II (синдром Хантера), тип III (синдром Санфилиппо), тип IV (синдром Моркио), тип VI (синдром Марото-Лами), тип VII (синдром Слая), тип IX (синдром Натовича).
Хотя клинические проявления мукополисахаридозов обычно выявляются по завершении неонатального периода, современные методы генетической диагностки зачастую позволяют выявлять эти лизосомные болезни уже у новорожденных. Вышеупомянутые типы МПС описаны ниже в соответсвующих рубриках данной главы.
В целях диагностики мукополисахаридозов осуществляется определение активности лизосомальных гидролаз, а также исследование мочи на содержание гликозаминогликанов (помимо клинических симптомов). При рентгенологическом исследовании у пациентов с мукополисахаридозами выявляются признаки раннего окостенения затылочно-теменного шва, так называемые «рыбьи позвонки».
Состояние, известное ранее как МПС тип VIII (синдром Ди Ферранте), предположительно вызываемый дефицитом фермента N-ацетилглюкозамисульфат-сульфатазы, который был описан в 1977 году, а по OMIM соответствует рубрике #253230, более в числе мукополисахаридозов не фигурирует. Рубрика в базе данных OMIM была сохранена с исторической целью.
- Читать "Синдром Ангельмана - причины, диагностика, лечение"
Оглавление темы "Детская неврология":- Синдром (болезнь) Лея у новорожденных - причины, диагностика, лечение
- Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) у новорожденных - причины, диагностика, лечение
- Дефицит короткоцепочечной ацил-КоА-дегидрогеназы (short-chain acyl-CoA dehydrogenase deficiency, SCADD) - причины, диагностика, лечение
- Дефект транспорта карнитина - причины, диагностика, лечение
- Дефицит среднецепочечной ацил-КоА-дегидрогеназы (medium-chain acyl-CoA dehydrogenase deficiency, MCADD) - причины, диагностика, лечение
- Дефицит длинноцепочечной 3-гидрокси-ацил-КоА-дегидрогеназы (long-chain 3-hydroxyacyl-СоА dehydrogenase deficiency, LCHADD) - причины, диагностика, лечение
- Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - причины, диагностика, лечение
- Недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (HMG-CoA SD) - причины, диагностика, лечение
- Мукополисахаридозы (МПС) у новорожденных - причины, диагностика, лечение
- Синдром Ангельмана - причины, диагностика, лечение