
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) у новорожденных - причины, диагностика, лечение
Окисление митохондриальных жирных кислот — это физиологическая реакция организма на голодание, инфекционный процесс и/или повышенную физическую нагрузку (с высокой мышечной активностью). Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) представляют группу наследственных нарушений, которые обычно проявляются у новорожденных и детей раннего возраста (хотя могут дебютировать у подростков и совершеннолетних индивидов).
Первый генетический дефект окисления жирных кислот описан в 1973 году (рабдомиолиз скелетных мышц и миоглобинурия). Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) являются нередкой патологией; по данным М. Gillingham и соавт., в некоторых популяциях они встречаются с частотой 1 случай на 9000 новорожденных.
Дефекты b-окисления митохондриальных жирных кислот — группа генетически детерминированных нарушений, вызванных недостаточностью функционирования специфических ферментов, расщепляющих коротко-, средне-, длинной/или очень длинноцепочечные жирные кислоты, что выражается в развитии катастрофических эпизодов голодания и/или индуцированной гиперкетотической гипогликемии, приводя к токси-ко-метаболическому поражению ЦНС и других органов и систем.
Всего известно около 20 ДОМЖК, для каждого из которых характерна недостаточность специфической ферментативной реакции в метаболизме жирных кислот, поступающих в организм алиментарным путем. P. Rinaldo и соавт. предлагают к рассмотрению классификацию основных ДОМЖК (из 13 разновидностей).
Нарушения бета-окисления длинноцепочечных ЖК (LCAD): а) дефект переносчика ДЦЖК, б) дефект транспорта карнитина.
Нарушения транспорта ЖК в митохондрии: а) недостаточность карнитин-пальмитоилтрансферазы I, б) недостаточность карнитин-пальмитоилтрансферазы II, в) недостаточность карнитин/ацилкарнитинтранслоказы.
Нарушения бета-окисления очень длинноцепочечных ЖК (VLCAD): а) недостаточность очень длинноцепочечной ацил-КоА-дегидрогеназы, б) недостаточность 3-функционального белка и в) изолированная недостаточность длинноцепочечной 3-гидроксиацилдегидрогеназы.
Нарушения бета-окисления среднецепочечных ЖК (MCAD): а) нарушения среднецепочечной ацил-КоА-дегидрогеназы ЖК, б) недостаточность среднецепочечной 3-кетоацил-КоА-тиолазы.
Нарушения бета-окисления короткоцепочечных ЖК (SCAD): недостаточность короткоцепочечной ацил-КоА дегидрогеназы.
Другие нарушения окисления ЖК: недостаточность митохондриальной 3-гидрокси- 3-метилглутарил-КоА-синтазы.
Нарушения кетогенеза: а) недостаточность 3-гидрокси-З-метилглутарил-КоА-лиазы, б) дефект дыхательной цепи, связанный с окислением ЖК (глутаровая ацидемия тип II).
Описаны клинически легкие и тяжелые фенотипы (варианты) ДОМЖК. Представим краткие данные о метаболизме жирных кислот.
Основные составляющие метаболизма жирных кислот сводятся к трем основным механизмам: 1) транспорт ЖК через плазматическую мембрану, 2) транспорт карнитина, 3) транспорт ЖК в митохондрии.
Длинноцепочечные жирные кислоты (ДЦЖК) принимают участие в синтезе фосфофолипидов, посттрансляционной белковой модификации, передаче клеточных сигналов, контролируют процессы транскрипции и оказывают влияние на проницаемость клеточных мембран. ДЦЖК — ключевой субстрат энергетического метаболизма (наибольшее значение имеют ЖК с длиной углеродной цепи С6 и С18, как насыщенные, так и моно-, ди-ненасыщенные). При истощении запасов гликогена (после голодания) триглицериды мобилизуются из жировых капель с высвобождением ЖК под действием липаз (на поверхности эндотелиальных клеток и гепатоцитов). Свободные ЖК транспортируются в клетки при участии тканеспецифичных белков. Не исключается возможность переноса ЖК посредством простой диффузии.
Высокая концентрация (внутриклеточная) карнитина, которая примерно в 50 раз превышает таковую в плазме крови, поддерживается системой активного транспорта. Для этой цели в сердечной и скелетных мышцах имеется транспортный белок, кодируемый геном OCTN2. В печени экспрессирован другой белок-транспортер с меньшей аффинностью.
ЖК активируются путем превращения в ацил-КоА-тиоэфиры на поверхности внешней митохондриальной мембраны под воздействием АТФ-зависимых синтетаз. Далее, под воздействием карнитин-пальмитоилтрансферазы I (CPTI), переносящей ацильные остатки ЖК от КоА к карнитину, происходит образование ацилкарнитиновых эфиров. Последние транспортируются через внутреннюю митохондриальную мембрану с участием карнитин-ацилкарнитинтранслоказы (CAT) — с заменой на свободный карнитин. Карнитин-пальмитоилтрансфераза II (СРТП) в митохондриальном матриксе переносит ацильные остатки ЖК от карнитина к КоА, в результате чего повторно образуются ацил-КоА-тиоэфиры. Карнитин не принимает участия в переносе коротко- и среднецепочечных ЖК (с длиной углеродной цепи <12 атомов) внутрь митохондрий.
CPTI является ферментом, лимитирующим скорость на стадии транспорта ЖК; на его активность влияет уровень малонил-КоА. Поступление углеводов в организм сопровождается повышением внутриклеточной концентрации малонил-КоА с подавлением активности CPTI и переключением метаболизма на синтез ЖК и триглицеридов.
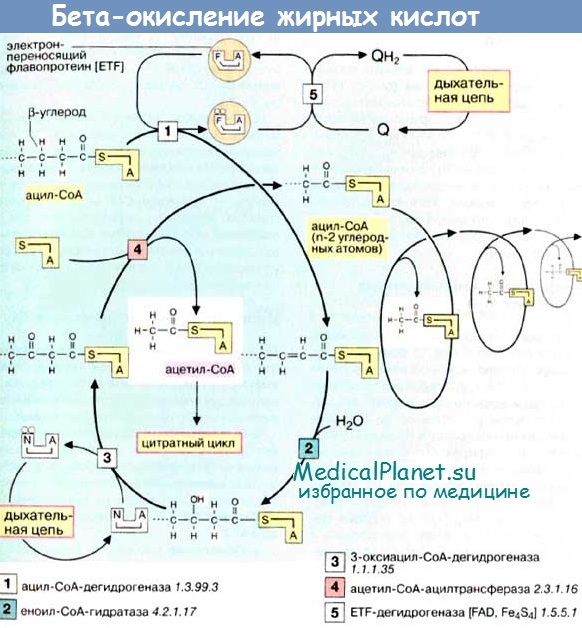
Процесс b-окисления жирных кислот и кетогенез
Окисление жирных кислот — метаболический процесс, в ходе которого происходят взаимодействие с кислородом (О2) и потеря молекулами ЖК атома водорода (Н). Процесс (3-окисления — последовательное отщепление одной молекулы ацетил-КоА от активированных ЖК под воздействием ферментов с различной специфичностью к длине цепи последних.
Первой реакцией в цикле b-окисления ЖК является дегидрогенизация, осуществляемая ацил-КоА-дегидрогеназами (содержат в качестве простетической группы флавинаденин динуклеотид — ФАД). В окислении ЖК принимают участие коротко- (SCAD), средне- (MCAD), длинно- (LCAD) и очень длинноцепочечная (VLCAD) ацил-КоА-дегидрогеназы. Три первые из описываемых дегидрогеназ относятся к ферментам митохондриального матрикса, а последняя (VLCAD) является энзимом, связанным с митохондриальной мембраной.
Окисление восстановленных флавопротеинов производится электрон-транспортирующим флавопротеином (ETF), который, в свою очередь, переправляет восстанавливающие эквиваленты другому флавопротеину — убихинон-оксидоредуктазе, локализованной на внутренней митохондриальной мембране. Последний переносит свои электроны в дыхательную цепь митохондрий через убихинон.
Второй реакцией в цепи b-окисления ЖК служит гидратация по двойной связи в положении 2,3, образовавшихся вследствие дегидрогенизации 2-транс-эноил-КоА в соответствующие L-3-гидроксиацил-КоА. Существуют две эноил-КоА-гидратазы (LHYD) функционирующая в составе мебраносвязанного b-функционального белка (TFP), а также кротоназа, гидратирующая средне- и коротокоцепочечные субстраты. В свою очередь, TFP является гетерооктамером, состоящим из а4b4-субъединиц, а-субъединица которого отвечает за LHYD-активность, а |3-субъединица — за 3-кетоацил-КоА-тиолазную активность (LKAT).
Третья реакция b-окисления ЖК заключается в дегидрогенизации 3-гидроксиацил-КоА в 3-кетоацил-КоА (с задействованием двух 3-гидроксиацилдегидрогеназ — для коротко- и средне-, а также длинноцепочечных субстратов соответственно, M/SCHAD и LCHAD). Третий фермент, отвечающий за 3-гидроксиацил-КоА-дегидрогеназную активность, представлен HAD-II.
Последняя стадия b-окисления ЖК — тиолитическое расщепление 3-кетоацил-КоА с образованием ацетил-КоА и соответствующего ацил-КоА-эфира с укороченной цепью, требующее участия трех ферментов (длинноцепочечная 3-кетоацил-КоА-тиолаза — LKAT, среднецепочечная 3-кетоацил-КоА-тиолаза — МКАТ, короткоцепочечная 3-кетоацил-КоА-тиолаза — SKAT).
Ацетил-КоА, сформированный в печени в период голодания в результате b-окисления ЖК, направляется в так называемую «цепь кетогенеза» (3-гидрокси-3-метилглутарил-КоА-синтетаза конденсирует 1 молекулу ацетил-КоА и 1 молекулу ацетоацетил-КоА, приводя к образованию 3-гидрокси-З-метилглутарил-КоА, который затем расщепляется гидрокси-3-метилглутарилКоА-лиазой с образованием ацетоацетата).
Ацетоацетат находится в состоянии равновесия с другим кетоновым телом — 3-гидроксибутиратом. Указанные кетоновые тела попадают в систему кровотока и доставляются тканям, где ацетоацетат переводится в активную форму — ацетоацетил-КоА (под воздействием ацетоацетат/ сукцинил-КоА-трансферазы). Затем ацетоацетил-КоА подвергается тиолитическому расщеплению и 2 молекулы ацетил-КоА вступают в цикл Кребса.
Таким образом, можно схематически представить 7 этапов бета-окисления ЖК с последовательным задействованием следующим ферментов:
• этап 1 — карнитин-пальмитоилтрансфераза I (CPTI),
• этап 2 — транслоказа,
• этап 3 — карнитин-пальмитоилтрансфераза II (СРТП),
• этап 4 — ацил-КоА-дегидрогеназа,
• этап 5 — эноил-КоА-гидратаза,
• этап 6 — 3-гидроксиацил-КоА-дегидрогеназа,
• этап 7 — 3-кетотиолаза.
Этиология/патогенез и генетические аспекты дефектов b-окисления митохондриальных жирных кислот (ДОМЖК)
Транспорт в митохондрии ДЦЖК с длиной углеродной цепи 12-18 требует участия L-карнитина, что не обязательно для коротко (С4-6) и среднецепоченых (С6-10) ЖК.
Карнитин-пальмитоилтрансфераза-1 (CPTI) формирует ацилкарнитиновые конъюгаты жирных кислот (этап 1), которые затем транспортируются в митохондрии транслоказой (этап 2). Оказавшись в митохондриальном матриксе, реакция связывания изменяется под воздействием карнитин-пальмитоилтрансферазы-2 (СРТП), содержащей свободные карнитин и длинноцепоченый ацил-КоА ЖК (этап 3).
Бета-окисление ацил-КоА ЖК происходит в процессе повторения 4-ферментного цикла, причем каждая его «спираль» сопровождается высвобождением одной молекулы ацетил-КоА (этапы 4-7).
Некоторые ДОМЖК, в частности дефицит среднецепочечной ацил-КоА (MCAD) и длинноцепочечной ацил-КоА (LCHAD) дегидрогеназ, связаны со специфическими мутациями в конкретных генах, вызывающими указанные болезни. Мутационный анализ считается менее сложным и дорогостоящим, чем исследование специфических ферментов, а кроме того, обладает 100% диагностической значимостью, если пациент является гомозиготным (по распространенной мутации).
Основным биохимическим признаком всех без исключения ДОМЖК является неадекватно низкая продукция кетонов из митохондриальных жирных кислот на фоне повышенных потребностей индивида в энергии. Вследствие этого возникает гипогликемия голодания, нередко ассоциированная с тяжелым (вторичным) ацидозом по причине недостаточного накопления продуктов интермедиарного обмена в ходе окисления ЖК.
Первый генетический дефект окисления жирных кислот описан в 1973 году. Распространенные мутации, связанные с ДОМЖК, ответственны не за все случаи указанных ферментопатий, но за большинство их них. В этой связи отрицательные результаты, полученные в ходе проведения специфического мутационного анализа, не позволяют полностью исключить те или иные варианты ДОМЖК, а требуют прямого исследования соответствующих ферментов.
Ген одной из изоформ CPTI (печеночной — L-CPTI) картирован на участке 11q13, а другой изоформы (мышечной — M-CPTI) — 22q13.3. В свою очередь, ген для СРТП картирован на локусе 1р32.
Ген, кодирующий фермент M/SCHAD (растворимый гомодимер митохондриального матрикса), располагается на участке 4q22-q26.
Гены обеих субъединиц TFP обнаруживаются на локусе 2р23.3-р24.1 (их активность регулируется сочетанно).
Все ДОМЖК передаются аутосомно-рецессивным путем.
Профилактика, прогноз и лечение дефектов b-окисления митохондриальных жирных кислот (ДОМЖК)
Профилактика ограничивается возможностями неонатального скрининга (тандемная масс-спектроскопия), которое рутинно в родильных домах РФ в настоящее время не осуществляется. Прогноз звисит от возраста дебюта варианта ДОМЖК, тяжести проявлений болезни и возможности проведения адекватной терапии. У детей, перенесших первоначальный «метаболический кризис», могут отмечаться рецидивирующие эпизоды декомпенсации, постепенно развиваться хронический неврологический дефицит в виде мышечной гипотонии, а также отставания в развитии.
Терапевтические возможности при ДОМЖК чрезвычайно малочисленны и по сути ограничиваются средствами симптоматического лечения, хотя существуют достаточно эффективные методы диетического лечения.
Клинические проявления и диагностика дефектов b-окисления митохондриальных жирных кислот (ДОМЖК)
Симптомы известных на сегодняшний день ДОМЖК сравнительно схожи при различных видах патологии этой группы и включают следующие проявления: тошнота, рвота, сомнолентность, печеночная энцефалопатия (последняя напоминает таковую при синдроме Рея и может приводить к коме или летальному исходу).
Синдром внезапной младенческой смерти (SIDS) вследствие метаболического кризиса при ДОМЖК констатируется у 30% детей с изначально тяжелыми эпизодами. Кардиомиопатия — еще одно частое, угрожающее жизни ребенка осложнение метаболической декомпенсации при многих ДОМЖК.
У части новорожденных, пораженных патологией описываемого спектра, могут полностью отсутствовать какие-либо симптомы ДОМЖК. Большая часть ДОМЖК диагностируется посредством исследования органических кислот в моче, а также содержания карнитина в плазме крови у детей с клиническими признаками синдрома Рея, кардиомиопатией, поражением печени неясного генеза, а также гипогликемией.
Поскольку каждый из дефектов b-окисления митохондриальных жирных кислот (ДОМЖК) ассоциирован со специфическим профилем содержания ацилкарнитина в плазме крови, в настоящее время в некоторых странах (США и др.) внедряются новые методы неонатального скрининга для выявления патологии описываемой группы уже при рождении.
Оглавление темы "Детская неврология":- Синдром (болезнь) Лея у новорожденных - причины, диагностика, лечение
- Дефекты b-окисления митохондриальных жирных кислот (ДОМЖК) у новорожденных - причины, диагностика, лечение
- Дефицит короткоцепочечной ацил-КоА-дегидрогеназы (short-chain acyl-CoA dehydrogenase deficiency, SCADD) - причины, диагностика, лечение
- Дефект транспорта карнитина - причины, диагностика, лечение
- Дефицит среднецепочечной ацил-КоА-дегидрогеназы (medium-chain acyl-CoA dehydrogenase deficiency, MCADD) - причины, диагностика, лечение
- Дефицит длинноцепочечной 3-гидрокси-ацил-КоА-дегидрогеназы (long-chain 3-hydroxyacyl-СоА dehydrogenase deficiency, LCHADD) - причины, диагностика, лечение
- Множественная недостаточность ацил-КоА-дегидрогеназы (multiple acyl-CoA dehydrogenase deficiency, MADD) - причины, диагностика, лечение
- Недостаточность 3-гидрокси-3-метилглутарил-КоА-синтазы (HMG-CoA SD) - причины, диагностика, лечение
- Мукополисахаридозы (МПС) у новорожденных - причины, диагностика, лечение
- Синдром Ангельмана - причины, диагностика, лечение