
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема развития болезней кости
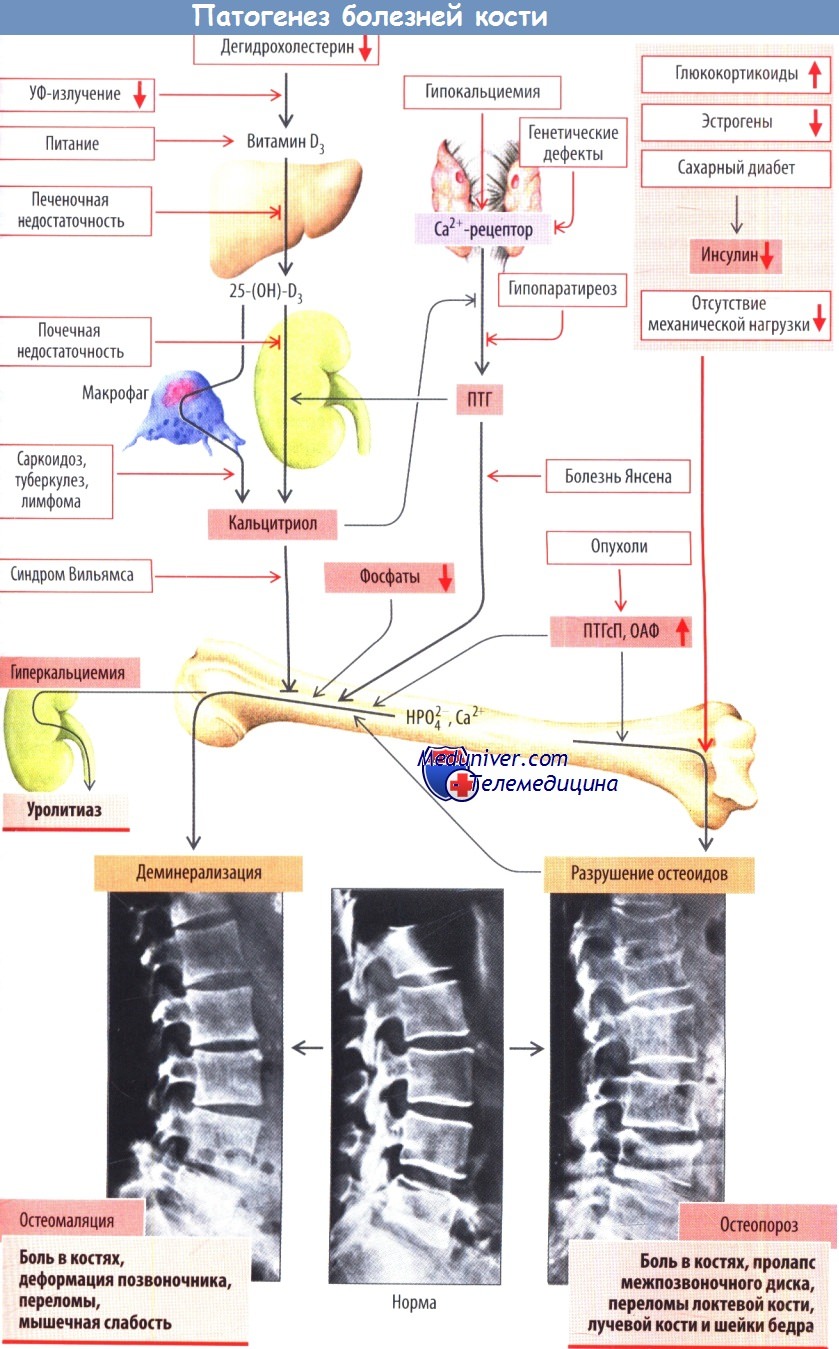
Нарушения обмена веществ в костях могут затрагивать костный матрикс или минерализацию.
Повышенная активность остеокластов с последующей стимуляцией остеобластов при болезни Педжета ускоряет жизненный цикл костной ткани с формированием структурно дезорганизованных костей, особенно подверженных деформациям и переломам.
Предполагаемые причины — повышенная чувствительность клеток-предшественниц остеокластов к 1,25(OH)2D3 или увеличение образования либо активности RANKL. Мутации гена RANKL приводят к похожей клинической картине. Ювенильная форма болезни Педжета обусловлена дефектом остеопротегерина.
Очень часто продолжительное нарушение баланса между формированием и резорбцией костной ткани заканчивается остеопорозом, уменьшающим плотность костей. Остеопороз может быть обусловлен избытком глюкокортикоидов, дефицитом эстрогенов (в период постменопаузы) и инсулина (сахарный диабет), диетой с низким содержанием кальция, курением и малоподвижностью (иммобилизация, тетраплегия, невесомость).
Однако чаще всего причина остеопороза остается неизвестной (первичный остеопороз). К проявлениям остеопороза относятся боль в костях даже в состоянии покоя и переломы костей (например, позвоночника, предплечья, шейки бедра). В крайних случаях может наблюдаться гиперкальциемия. В зависимости от этиологических факторов остеопороз может быть локальным (например, под гипсовой повязкой) или генерализованным (например, из-за избытка глюкокортикоидов).
При остеомаляции и рахите нарушается минерализация костного матрикса (остеоида) или зоны роста. Нарушения, возникшие до завершения роста в длину и до наступления эпифизарного слияния, приводят преимущественно к рахиту (расширение зоны роста и нарушенный рост). Таким образом, гипофосфатемия способствует выживанию хондроцитов в зонах роста.
После того как рост кости в длину прекращается, ослабленная минерализация недавно сформированного остеоида (сформированного в ходе нормального ремоделирования кости) вызывает остеомаляцию. Как рахит, так и остеомаляция могут быть обусловлены пониженным образованием кальцитриола, например, при недостаточном поступлении с пищей или нарушении всасывания витамина D в кишечнике в сочетании с отсутствием воздействия УФ-излучения при печеночной недостаточности, дефиците эстрогенов (постменопауза) или хронической почечной недостаточности.
Гипофосфатемия (фосфатный диабет, синдром Фанкони) или хронический почечный канальцевый ацидоз даже без дефицита кальцитриола могут вызывать остеомаляцию.
Проявления рахита: задержка роста, саблевидные голени или деформация коленного сустава с образованием угла между голенью и бедром, открытого кнаружи, деформации позвоночного столба, муфтообразные реберно-хрящевые сочленения (рахитические четки), а также тонкие и мягкие, особенно затылочная, кости черепа (краниотабес). Остеомаляция сопровождается болью в костях (боль при движении), появлением светлых полос деминерализация на рентгенограммах (псевдопереломы, или зоны Лоозера) и мышечной слабостью (дефицит Са2+).
Деминерализация костей может увеличивать почечную экскрецию Са2+ и фосфатов и, как следствие приводить к мочекаменной болезни. Стимулировать резорбцию костей способны опухоли (образование ПТГсП и ОАФ). При первичном гиперпаратиреозе за счет бесконтрольной пролиферации ПТГ продуцирующих клеток нормальная костная ткань замещается фиброзной.
Нарушения формирования и резорбции костей могут быть результатом генетических дефектов например вследствие мутации гена коллагена I типа (несовершенный остеогенез) или мутации, вызывающей инактивацию CBFA1 [ключично-черепная дисплозия). Дефект щелочной фосфатазы (гипофосфатазия) ослабляет минерализацию костей. Дефект субъединицы TC1RG1 Н+-насоса, Cl--канала CICN7, CAI или RANK нарушает функцию остеокластов [остеопе: роз). Резорбция костей ослабляется генетическим де фектом протеазы катепсина К (пикнодизостоз, воз можно, болезнь Тулуз-Лотрека).
Генетические дефекты Са2+-чувствительного рецептора приводят к семейной доброкачественной гиперкальциемии, мутациям, активирующим ПТГ-рецепторы при болезни Янсена (гиперкальциемия гипофосфатемия, мальформация скелета, карликовость). Генетические дефекты, нарушающие выделение ПТГ (гипопаратиреоз) или действие ПТГ (псевдогипопаратиреоз, например, за счет дефектного G-белка), сопровождаются гиперкальциемией и, отчасти, мальформациями костей. Более того, наследственный дефицит ПТГ может приводить к кальцификации и последующему повреждению базальных ядер.
Генетический дефект 1а-гидроксилазы ведет к псевдодефицитному витамин D-зависимому рахиту, наследственное повышение чувствительности к кальцитриолу — к гиперкальциемии при синдроме Вильямса.
Существует множество вариантов редких генетических дефектов (например, FGF3), которые нарушают образование хрящевой ткани (остеохондродисплазия).
- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Патогенез заболеваний в схемах":- Схема развития болезней почек при беременности (нефропатии беременных)
- Схема развития гепаторенального синдрома
- Схема развития мочекаменной болезни (МКБ)
- Схема развития нарушения обмена воды - гипергидратации и дегидратации
- Схема развития нарушения обмена калия - гиперкалиемии и гипокалиемии
- Схема развития нарушения обмена магния - гипермагниемия и гипомагниемия
- Схема развития нарушения обмена кальция - гиперкальциемии и гипокальциемии
- Схема развития нарушения обмена фосфатов - гиперфосфатемии и гипофосфатемии
- Схема образования и разрушения кости
- Схема развития болезней кости