
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Ваарденбурга - причины, клиника, диагностика
Синдром Ваарденбурга (СВ), описанный в 1951 г.голландскм врачом Петрусом Ваарденбургом, служит прототипом врожденного нарушения пигментации. Хотя первоначально он был описан как синдром, сочетающий дефекты пигментации волос (полиоз или белую прядь) и радужной оболочки, врожденную глухоту и черепно-лицевые пороки развития, впоследствии выяснилось, что в этот синдром могут входить и другие фенотипические проявления. Было описано четыре типа синдрома Ваарденбурга, от СВ1 до СВ4.
Открытие молекулярных мутаций, влияющих на различия в типах СВ, помогло объяснить широкий спектр проявлений, а также вывить важность специфических генов для развития различных тканей и органов. Хотя практически во всех случаях СВ1 и СВ3 наблюдается мутация ДАХ3, у лиц с СВ4 присутствуют или гомозиготные мутации EDN3 (гена эндотелина-3), или EDNRB (гена рецептора эндотелина В), или гетерозиготные мутации SOX10. Что касается СВ2, то этот тип синдрома возникает более гетерогенным путем, поскольку мутации в MITF обнаружены лишь у немногих пациентов с СВ2.
Дифференциация врожденных заболеваний с нарушением пигментации:
- Синдром Ваарденбурга типов 1-4
- Синдром Титце
- Пиебалдизм
- Синдром Вульфа
- Генерализованное витилиго
- Сегментарное витилиго
- Синдром Вогта-Коянаги-Харада
- Химическая лейкодерма
- Туберозный склероз (гипопигментированные пятна и бляшки)
- Синдром Ципрковски-Марголиса (синдром Х-сцепленого альбинизма и глухоты)

и dystopia canthorum (латеральное смещение медиального угла глазной щели).
а) Синдром Ваарденбурга типа 1. Пациенты с СВ1 (OMIM #193500) обычно бывают гетерозиготными по мутациям в РАХ3, следовательно, СВ 1 наследуется по аутосомно-доминантному типу. Хотя с СВ1 ассоциируется много различных мутаций в РАХ3, все эти мутации либо являются функционально нулевыми аллелями, либо аннулируют взаимодействие РАХЗ с ДНК.
У пациентов с СВ1 аномалии пигментации сопутствуют черепно-лицевым деформациям. Dystopia canthorum представляет собой латеральное смещение медиального угла глазной щели и является ведущим признаком черепно-лицевой деформации, которая присутствует практически во всех случаях СВ1. С синдромом СВ1 ассоциируются также другие черепно-лицевые аномалии, такие как расширенная переносица, гипоплазия крыльев носа и синофриз (увеличение и сращение бровей). Полиоз, то есть наличие белой пряди волос в лобной области — самая распространенная аномалия пигментации для данного типа синдрома.
Белые пятна депигментации на коже встречаются реже и часто расположены по вентральной срединной линии, что отражает нарушение миграции дисфункциональных прекурсоров меланоцитов из места их происхождения в дорсальной части неврального гребня. Пигментные аномалии радужных оболочек, в том числе полная гетерохромия (радужки разного цвета), частичная гетерохромия (вариации цвета в пределах радужки) или гипопластические голубые радужки, также могут быть ассоциированы с СВ1. Кроме того, с синдромом СВ1 может сочетаться преждевременное поседение волос. Врожденная глухота отмечается в 57% случаев.
Дефекты пигментации при СВ1 объясняются ролью РАХЗ в экспрессии MITF с последующим воздействием на выживание меланоцитов в ходе развития. Черепно-лицевые аномалии при СВ1 связаны с важностью РАХЗ для процесса управления развитием костных и хрящевых структур лица, происходящих из нервного гребня, особенно тех, что способствуют формированию лобной кости. Нейросенсорная глухота при не полной пенетрации в случае СВ1 развивается вследствие вариабельной неспособности меланобластов мигрировать или выживать в stria vascularis латеральной стенки улитки.
б) Синдром Ваарденбурга типа 2. Генетический анализ сцепления у семей с СВ2 (OMIM #193510, 608890, 600193, 606662) идентифицировал локус MITF в качестве кандидатного гена заболевания. В семьях с СВ2 было обнаружено как минимум девять различных мутаций в кодирующем регионе гена MITF. Однако мутациями MITF объясняется лишь незначительная часть случаев СВ2 (15%). С типом СВ2 ассоциируется также мутация в гене фактора транскрипции SLUG/SNAI2, а кроме того — гетерозиготные делеционные мутации в гене SOX10, однако в будущем будут, вероятно, обнаружены мутации в других, пока еще не установленных генах.
Синдром Ваарденбурга типа 2 (СВ2) наследуется по аутосомно-доминантному типу. Поскольку большинство из известных при СВ2 мутаций MITF изменяют регион HL-HZip, нарушая тем самым димеризацию мутантного MITF с MITF дикого типа, патологическое развитие пигментации в большинстве случаев происходит, вероятно, вследствие гаплонедостаточности (уменьшение дозы генного продукта, экспрессии или активности белка), а не в результате доминантно-негативных изменений. Вследствие доминантно-негативных эффектов мутантного MITF развивается, вероятно, описанный ниже синдром Титце.
Тот факт, что мутации в MITF являются причиной одного из типов СВ, привлекателен с механистической точки зрения, учитывая исключительную важность MITF для выживания меланоцитов в процессе развития. В ходе эмбриогенеза, в процессе развития меланоцитов, между РАХ3, SOX10 и MITF существуют эпистатические взаимодействия. РАХ3 и SOX10, будучи транскрипционными факторами, синергически трансактивируют MITF. Поскольку мутации РАХ3 и SOX10 также вызывают симптомы со стороны органов слуха и нарушения пигментации при других типах СВ, то вероятно и дефект трансактивации MITF является определяющим фактором в нарушении развития пигментных клеток при этих типах синдрома.
SLUG/SNAI2 могут действовать в качестве транскрипционной мишени в том же сигнальном пути, что и важный, расположенный ниже по ходу транскрипции эффектор MITF в меланоцитах, активность которого блокируется при SLUG-ассоциированном СВ2.
Хотя пигментные аномалии кожи и радужных оболочек, а также возможность потери слуха наблюдаются при всех типах синдрома Ваарденбурга, тип СВ2 отличается тем, что ограничен наличием только этих симптомов. Диагностические критерии СВ2 уже определены. Диагноз СВ2 устанавливается лицам, которые удовлетворяют двум из следующих четырех критериев в отсутствие dystopia canthorum, деформации конечностей или болезни Гиршпрунга:
1. Врожденная нейросенсорная потеря слуха.
2. Пигментные аномалии радужной оболочки глаза
а. Полная гетерохромия радужных оболочек (глаза разного цвета).
б. Частичная или сегментарная гетерохромия (сегменты голубой или коричневой пигментации в одном или обоих глазах).
в. Гипопластические голубые глаза (характерный бриллиантово-голубой цвет обоих глаз).
3. Пигментные аномалии волос.
а. Белая прядь волос в лобной области с рождения или в подростковом периоде.
б. Преждевременное поседение до 30 лет.
4. Родственники первой или второй линии с двумя или более критериями с 1 по 3.
Обзор 124 случаев СВ2 и 270 случаев СВ1 выявил различия в фенотипической пенетрации между двумя типами синдрома. Соответственно были зарегистрированы: нейросенсорная потеря слуха у 77% и 57% пациентов, гетерохромия радужных оболочек у 48% и 27%, гипопластические голубые глаза у 9% и 17%, раннее поседение у 23% и 26% и белые пятна на коже у 6% и 31% пациентов. Более высокая частота нейросенсорной потери слуха при СВ2 обусловлена, вероятно, трудностями в установлении диагноза СВ2 у лиц без потери слуха.
Потеря слуха является врожденной, нейросенсорной, не прогрессирует и значительно варьирует как между семьями, так и в пределах одной семьи. В серии из 81 случая СВ2 у 84% пациентов отмечалась двухсторонняя потеря слуха, у 40% — глубокая потеря слуха с одной или с обеих сторон.
в) Синдром Ваарденбурга типа 3. СВ3 (OMIM #148820), известный также как синдром Кляйна-Ваарденбурга, считается вариантом СВ 1. Большинство пациентов являются гетерозиготными по мутации в РАХЗ, хотя было описано и несколько гомозигот с тяжелым поражением. В отличие от фенотипа СВ1, с фенотипом СВЗ не коррелируют никакие специфические мутации в РАХЗ, за возможным исключением миссенс-мутации в Asp47, хотя либо при гомозиготных, либо при сочетанных гетерозиготных мутациях гена РАХЗ может реализоваться фенотип тяжелой формы СВ3, вместо СВ1.
В дополнение к признакам СВ1 у пациентов с СВ3 отмечаются мышечно-скелетные аномалии, которые проявляются в виде контрактур суставов и гипоплазии мускулатуры конечностей. Фенотип СВЗ подтверждает ранее описанную роль РАХЗ в активации факторов транскрипции, которые определяют развитие мышц и костей конечностей, отличную от его роли в качестве регулировщика развития структур, происходящих из неврального гребня.
г) Синдром Ваарденбурга типа 4. СВ4 (OMIM #277580), известный также как синдром Шаха-Ваарденбурга, вызывается гетерозиготными мутациями в гене фактора транскрипции SOX10 или гомозиготными мутациями в гене, кодирующем пептидный лиганд эндотелина-3, EDN3, или его рецептор, EDNRB3.
Кроме управления некоторыми аспектами развития меланоцитов, эти гены являются важными детерминантами развития клеток нервной системы, иннервирующих дистальные отделы ободочной кишки, чем объясняется ассоциация с болезнью Гиршпрунга (мегаколон). СВ4 представляет собой комбинацию фенотипа СВ1 с болезнью Гиршпрунга или врожденным аганглиозом ободочной кишки. СВ4 является комбинаццией фенотипа СВ1 с болезнью Гиршпрунга.
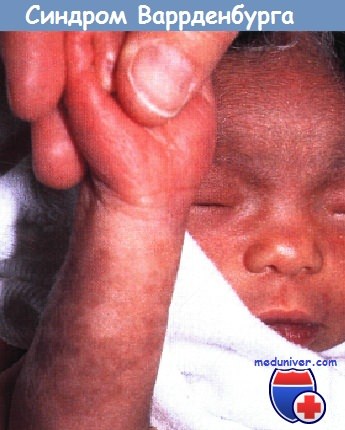
Обратите внимание на белую прядь волос, характерную для пьебалдизма.
Имеется семейный анамнез подобных кожных очагов в сочетании с утратой слуха и офтальмологическими признаками, характерными для синдрома Ваарденбурга.

- Рекомендуем далее ознакомиться со статьей "Синдром Титце - причины, клиника, диагностика"
Оглавление темы "Альбинизм и врожденные нарушения пигментации.":- Эпидемиология альбинизма и врожденных нарушений пигментации
- Причины и признаки кожно-глазного альбинизма типа 1 (КГА1)
- Причины и признаки кожно-глазного альбинизма типа 2 (КГА2)
- Причины и признаки кожно-глазного альбинизма типа 3 (КГА3)
- Синдром Германски-Пудлака - причины, клиника, диагностика
- Синдром Чедиака-Хигаши (СЧХ) - причины, клиника, диагностика
- Синдром Ваарденбурга - причины, клиника, диагностика
- Синдром Титце - причины, клиника, диагностика
- Пьебалдизм и симметричный дисхроматоз - причины, клиника, диагностика
- Прогноз и лечение альбинизма