
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Симптомы и клиника кератодермий с преобладанием кожных признаков
а) Точечная кератодермия. Точечная кератодермия (синдром Бушке-Фишера-Брауера; MIM 146800) обычно наследуется по аутосомно-доминантному признаку. В отличие от большинства других наследственных ЛПК, точечная ЛПК появляется во взрослом возрасте в форме многочисленных кератинизированных папул. Её могут обнаружить случайно, хотя подошвенные очаги могут вызывать значительные функциональные ограничения, а также приводить к развитию вторичной очаговой кератодермии.
На основании постепенного скопления дискретных индивидуальных очагов можно сделать вывод о наличии дефекта в одном унаследованном аллеле и об экспрессии заболевания в случае повреждения оставшегося аллеля. В поколениях нескольких семей сообщалось об ассоциации со злокачественными заболеваниями почек, желудка, грудной железы и толстого кишечника, но в целом такая ассоциация не подтверждена. В любом случае, это состояние, вероятно, генетически гетерогенное.
Размер папул в разных семьях варьирует от нескольких миллиметров до крошечных нитевидных очагов (кератодермия по типу «валика музыкальной шкатулки»). Подобным образом и гистология очагов может иметь картину ортокератотического или паракератотического гиперкератоза. Предполагается участие как минимум двух генных локусов, хотя наиболее вероятен участок 15q22.2-15q22.31. К другим клинически похожим состояниям относятся точечная кератодермия ладонных складок и очаговый акральный гиперкератоз краев ладоней и подошв с наличием или отсутствием эластоидоза.
Папулезные ладонно-подошвенные очаги могут наблюдаться при других генодерматозах, особенно при болезни Дарье, при которой точечные углубления, типичные в молодом возрасте, позднее становятся папулезными. Приобретенные случаи точечных ладонно-подошвенных очагов наблюдаются при диоксиновой токсичности и хроническом отравлении мышьяком (арсеницизме).
б) Эпидермолитическая кератодермия. Примером неосложненной диффузной кератодермии является эпидермолитическая ЛПК (ЭЛПК, синдром Фернера [Vorner]; MIM 144200). ЭЛПК, вероятно, представляет собой самую распространенную ЛПК и наследуется по аутосомно-доминантному признаку вследствие дефекта кератина 9, специфического кератина для кожи ладоней и подошв. Заболевание проявляется в детстве в форме диффузной ЛПК с четким ливидным переходом в нормальную кожу по краям ладоней и подошв.
В тяжелых случаях образуются пузыри, хотя у взрослых хрупкость эпидермиса чаще проявляется в форме трещин. Гистологическая картина шиповатого слоя демонстрирует вакуолизированные кератиноциты, со скоплениями кератиновых филаментов на электронно-микроскопическом уровне, в сочетании с ортогиперкератозом рогового слоя. Дефекты в кератине 1, предполагаемом партнере кератина 9 в ладонно-подошвенной коже, также вызывают диффузную трансгредиентную кератодермию с эпидермолитическим гиперкератозом на других участках кожи, но экстраплантарное поражение может быть слабо выраженным.
Специфический дефект домена 1В в кератине 1 приводит к формированию в некоторых поколениях трубчатых тонофиламентных структур. Кератодермия может быть также признаком тяжелого буллезного эпидермолиза типа Доулинга-Меара (MIM 131760;) вследствие мутаций кератинов базального слоя 5 и 14. Механизмы, посредством которых дефекты в генах кератинов вызывают ладонно-подошвенный гиперкератоз, обсуждаются выше.
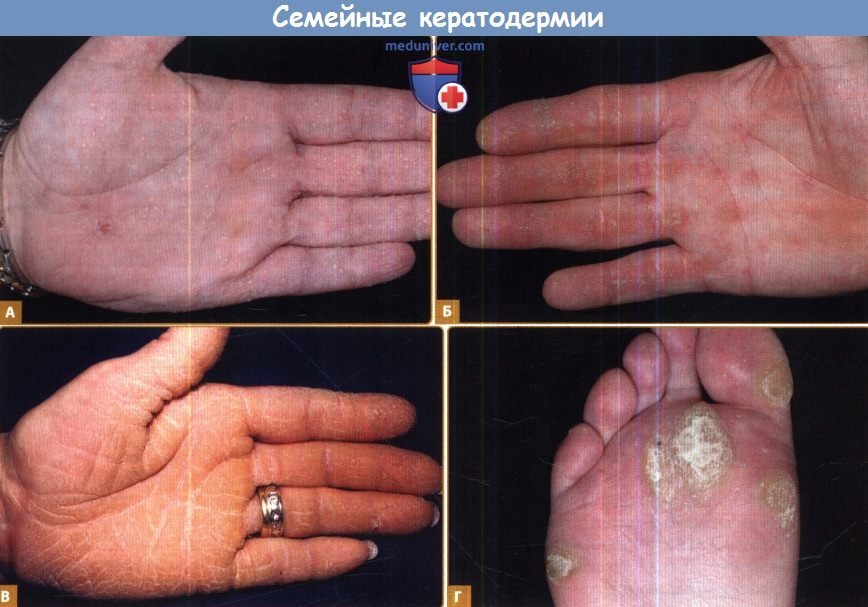
А. Точечная, которая обычно не появляется до взрослого возраста. Очаги четче выделяются при погружении ладоней на несколько минут в воду.
Б. Полосовидная, часто вследствие десмосомальных нарушений.
В. Диффузная, в данном случае с трещинами и четкой демаркацией, характерной для дефектов кератина 9.
Г. Очаговая, наблюдается в качестве изолированного нарушения в результате мутаций в кератине 6с.
На практике различие между этими формами не всегда четко выражено, особенно на коже ладоней
в) Неэпидермолитическая диффузная ладонно-подошвенная кератодермия (ЛПК). Неэпидермолитическая диффузная ЛПК, ограниченная кожей ладоней и подошв, имеет гетерогенную природу. От термина «синдром Унны-Тоста» (М1М 600962) следует, вероятно, отказаться, поскольку члены семьи Тоста фактически имели эпидермолитическую ЛПК. В поколениях одной из семей диффузный неэпидермолитический фенотип наблюдался вследствие дефекта в вариабельном участке гена, кодирующего кератин I, а в другой семье этот фенотип был картирован на локусе, включающем генный кластер кератинов типа II.
Был идентифицирован еще один локус, расположенный проксимально к генному кластеру кератинов типа II.
г) Очаговая ладонно-подошвенная кератодермия (ЛПК). Очаговая ЛПК может осложнять точечную ЛПК на стопах или развивается отдельно. В последнем случае её трудно отличить от физиологических омозолелостей, тенденция к образованию таких омозолелостей, действительно, может иметь генетическую основу. Раннее начало очаговой кератодермии, какие бы причины её не вызвали, приводит к утрате трудоспособности («болезненные наследственные омозолелости»), причем поражение бывает настолько сильным, что приковывает пациентов к инвалидной коляске.
Аутосомно-доминантное наследование очаговой ЛПК обычно ассоциируется с дистрофией ногтей и поражением слизистых оболочек как частью спектра пахионихии, но даже в отсутствие дистрофии ногтей поражение вызывается мутацией в кератине 16. В большинстве случаев изолированной очаговой ЛПК генетические причины не идентифицированы, но в трех семьях она была вызвана мутацией в кератине 6с. Болезненная очаговая ЛПК наблюдается также при тирозинемии типа II.
д) Полосовидная ладонно-подошвенная кератодермия (ЛПК). Полосовидная ЛПК (MIM 148700) — характерный вариант очаговой ЛПК, при котором отмечается выраженное линейное поражение ладонных поверхностей пальцев и менее очевидное поражение соответствующих участков подошв. Гистологическая картина аутосомно-доминантно наследуемой полосовидной ЛПК демонстрирует незначительное расширение межклеточных пространств. Большинство случаев вызваны дефектами в гене, кодирующем белки десмосомальных бляшек. Чаще всего поражается десмоглеин 1 (DSG1), но сообщалось также о мутациях в десмоплакине 1.
Считается, что кератодермия вследствие дефектов в этих генах вызвана гаплонедостаточностью, то есть пониженные количества релевантного структурного белка, экспрессированного в десмосомальной бляшке приводит к механической слабости, которая проявляется в точках максимального стресса. Однако как и в случае с кератинами, десмосомальные компоненты участвуют также в межклеточной сигнализации и регулировке роста. Полосовидная ЛПК может также наблюдаться в случае мутаций в кератине I, и, действительно, полосовидность является неспецифическим компонентом диффузных кератодермий легкой степени.
Большинство случаев аутосомно-доминантной полосовидной ЛПК простые, но дефекты в десмоплакине приводят к ряду синдромов с кардиомиопатией и шерстистыми волосами.
е) Лорикриновая кератодермия. Лорикриновая кератодермия (синдром Камисы [Camisa];MIM 640117) была описана как вариант синдрома Фовинкеля. Для этого варианта характерна трансгредиентная кератодермия с похожей узорчатой картиной «пчелиных сот». Трансгредиентная кератодермия приводит к круговому поражению пальцев, рубцующимся перетяжкам и иногда к автоампутации. Кроме того, сообщается о легкой степени генерализованного ихтиоза и о коллодиевом плоде или генерализованной десквамации при рождении. По сравнению с истинным синдромом Фовинкеля вследствие мутаций в Сх26, края кератодермии на запястьях диффузные и отсутствует глухота.
Сообщается о нескольких мутациях, наиболее частая мутация 730insG обнаружена в семьях из Великобритании, Японии, Германии и Италии. Во всех случаях это одиночные нуклеотидные включения, приводящие к сдвигу рамки считывания и запаздыванию стоп-кодона на 22 аминокислоты. В удлиненном карбокси-терминальном домене многие глициновые остатки замещаются аргинином, что резко изменяет свойства лорикринового пептида путем генерации сигнала ядерного распознавания (см. выше).
ж) Синдром Урье (Huriez). Синдром Урье (ЛПК со склероатрофией, склеротилозом; MIM 181600), аутосомно-доминантное заболевание, характеризуется диффузной ЛПК, которая поражает главным образом кожу ладоней. Вызвавший нарушение генный дефект неизвестен, но сообщалось о связи с хромосомой 4q23. Типичны склеродактилия, брахидактилия эритема и/или атрофия кожи, а также различные изменения ногтей; сообщалось также о гипогидрозе. В пораженной коже был идентифицирован дефицит клеток Лангерганса.
Данное состояние предрасполагает к развитию на пораженной коже плоскоклеточных карцином, которые появляются в третьей или четвертой декаде жизни и необычно склонны к метастазам. Причинная терапия отсутствует, но сообщалось о благоприятном эффекте системных ретиноидов.
з) Болезнь острова Меледа. Болезнь острова Меледа (MIM 248300) — редкое аутосомно-рецессивное заболевание, впервые описанное у жителей прибрежных районов Средиземноморья. Трансгредиентный (т.е. распространяющийся за латеральный край ладоней и проксимально за запястья) гиперкератоз восковидный, цвета слоновой кости или желтого, обычно воспаленный или мацерированный, с ливидной границей. Дерматофитная суперинфекция, к которой склонны многие кератодермии, может имитировать такую картину.
Характерны околосуставные подушечки на пальцах, подобные восковидные очаги на акральных участках, ангулярный хейлит и круговые склерозирующие и рубцующиеся очаги на пальцах; неудивительно, что ногти часто дистрофичные. Это типичное для региона Средиземноморья заболевание возникает вследствие мутаций в гене ARSB, кодирующем SLURP-1, аналог ацетилхолинового рецептора.
и) Другие трансгредиентные кератодермии. Сообщалось о рецессивных трансгредиентных/мутили-рующих заболеваниях с фенотипом, напоминающим болезнь острова Меледа, но генетически отличными от неё. Описано очень редкое аутосомно-рецессивное заболевание (KLICK-синдром), при котором рубцующаяся ЛПК сочетается с ихтиозом и причудливой формы полосовидным гиперкератозом сгибательных поверхностей. Недавно было установлено, что это нарушение возникает вследствие дефекта в гене POMP, который кодирует белок, являющийся шапероном для созревания протеазы.
Были описаны также случаи аутосомно-доминантно наследуемых трансгредиентных ЛПК, для которых применялся термин синдром Грейтера (Greither), хотя эти случаи, возможно, не являются одной нозологической единицей. Одни аутосомно-доминантные кератодермии трансгредиентного спектра возникают в результате мутаций в кератине 1, другие, например синдром Сайберт (Sybert), не связаны с кератиновыми нарушениями.
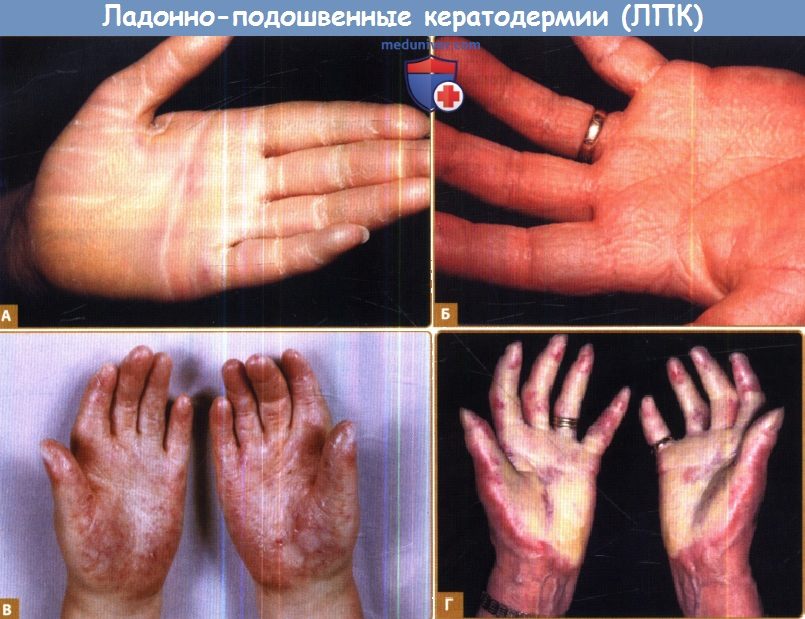
А. Узорчатая картина («пчелиные соты») лорикриновой кератодермии; обратите внимание на круговую кератодермию и формирующиеся констрикторные перетяжки.
Б. Сливающиеся папулы при ЛПК Фовинкеля с нарушением слуха вследствие мутаций коннексина 26; очаги в форме пчелиных сот и круговая ЛПК с констрикторными перетяжками также наблюдаются при этом синдроме.
В. Синдром Урье (склеротилоз), чреватый высоким риском плоскоклеточной карциномы (приводится с разрешения Dr. Cameron Kennedy, Bristol Royal Infirmary, Великобритания.)
Г. Болезнь острова Меледа вследствие мутаций в ARS, компонент В, с восковидным гиперкератозом, склеродактилией и констрикторными перетяжками.
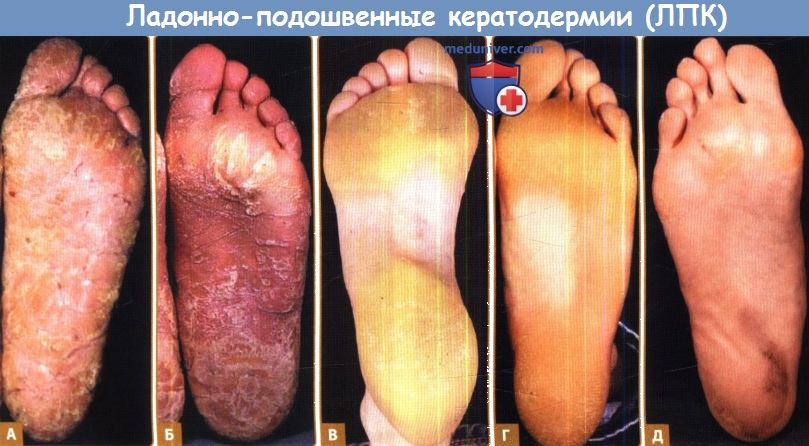
А. Диффузная ЛПК с трещинами вследствие мутации в кератине-1 и только с локализованным эпидермолитическим гиперкератозом в области локтей, коленей и сгибательных поверхностей.
Б. Подобные изменения при синдроме Папийона-Лефевра вследствие мутации в катепсине С (приводится с разрешения Barts и London NHSTrust, Великобритания).
В. Синдром Хоуэла-Эванса (ЛПК с семейной карциномой пищевода): не полностью сливающаяся кератодермия, не поражающая подъем стопы.
Г. Похожие изменения вследствие мутации включения в кератин 1.
Д. Очаговая ЛПК ч нарушением слуха вследствие мутации A7445G в митохондриальной ДНК.
- Рекомендуем далее ознакомиться со статьей "Симптомы и клиника кератодермии с эктодермальной дистрофией"
Оглавление темы "Ладонно-подошвенные кератодермии (ЛПК).":- Причины и механизмы развития ладонно-подошвенной кератодермии (ЛПК)
- Клиническая классификация ладонно-подошвенной кератодермии (ЛПК)
- Симптомы и клиника кератодермии с эктодермальной дистрофией
- Симптомы и клиника кератодермии с поражением слизистых
- Симптомы и клиника кератодермии с поражением сердца
- Симптомы и клиника кератодермии с нарушением слуха
- Симптомы, клиника кератодермии с невропатией и задержкой речевого развития
- Анализы и биопсия при кератодермии
- Дифференциальная диагностика кератодермии
- Современное лечение кератодермии