
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Симптомы и клиника кератодермии с эктодермальной дистрофией
а) Врожденная пахионихия (ВП). Врожденная пахионихия (ВП) представляет собой спектр доминантно наследуемых заболеваний, ассоциированных с очаговой ЛПК. Кроме болезненной очаговой ЛПК и гипертрофической дистрофии дистальных участков ногтей, имеются многочисленные другие ассоциации. Клинически выделяют два основных синдрома, при которых различные сочетанные признаки могут вызывать значительную дополнительную морбидность и приводить к нетрудоспособности. ВП-1 (Ядассона-Левандовского; MIM 167210) сочетается с фолликулярным кератозом, лейкокератозом полости рта и охриплостью голоса.
Некоторые очаги в полости рта могут напоминать кандидоз слизистых оболочек, а поражение гортани может вызвать у младенцев обструкцию дыхательных путей. Более редкая ВП-2 (Джексона-Лоулера; MIM 167210) характеризуется многочисленными сально-волосяными кистами, наличием зубов при рождении и изменениями волос, в частности торчащими бровями. Обширные и инфицированные кисты в складках вульвы и мошонки могут проявляться как гнойный гидраденит. ВП-1 типично ассоциируется с мутациями в KRT6A или KRT16, a ВП-2 — смутациями в KRT6B или KRT17, что отражает тип экспрессии соответствующих кератинов: К16, например, является основным вторичным кератином в эпителии полости рта и гениталий.
Тяжесть заболевания варьирует как между семьями, так и в одной семье, а мутации в одинаковых генах могут приводить к неполным фенотипам, таким как ЛПК без дистрофии ногтей или множественной стеатоцистоме без ЛПК.
Однако детальный анализ пациентов с мутациями KRT16 указывает на то, что мутации, оказывающие более разрушительное воздействие на структуру белка К16, приводят к развитию более тяжелого фенотипа. Недавнее крупное исследование породило сомнения в постоянстве сочетаний клинического синдрома и мутировавшего кератина, и было предложено обозначать врожденную пахионихию (ВП) с указанием пораженного кератина, например ВП-6а, ВП-6b и т.д. и ВП-Н (неизвестен). Целью генотипической классификации является индивидуализированная целенаправленная молекулярная терапия, уже имеющая хорошие перспективы при этом заболевании.
б) Синдром Шепша-Шульца-Пассаржа. Синдром Шепфа-Шульца-Пассаржа (MIM 227450) является аллельным вариантом онихо-одонтодермальной дисплазии (MIM 257980), спектра аутосомно-рецессив ных эктодермальных дисплазий, основной фенотип которых проявляется аномалиями ногтей, олигодонтией с аномалиями зубов и гипотрихозом. Аномалии зубов могут развиваться у гетерозигот. К дополняющим ладонно-подошвенную кератодермию признакам относятся эритематозные очаги на лице и атрофия сосочков языка. При данном синдроме у пациентов обычно развиваются доброкачественные опухоли придатков кожи.
Характерны апокринная гидроцистома в форме кист на веках, наблюдаются также эккринная порома и сирингофиброаденома. Сообщается о плоскоклеточных и базальноклеточных карциномах, а также порокарциноме на пораженной коже. Нарушения в спектре онихо-одонтодермальных дисплазий возникают в результате мутаций в WNT10a. Белок, кодированный этим геном, является членом семейства секретируемых сигнальных молекул, которые блокируют деградацию β-катенинового комплекса и, следовательно, позволяют ему модулировать экспрессию гена.
WNT-сигнализация играет важную роль во многих механизмах развития, a WNTlOa участвует в морфогенезе зубов и волосяных фолликулов, а также в формировании эктодермальных гребней.
в) Синдром Олмстеда. Синдром Олмстеда (мутилирующая ладонно-подошвенная кератодермия с периорифициальными бляшками) является тяжелым заболеванием. Врожденная диффузная, четко отграниченная и прогрессирующая кератодермия ладоней и подошв вызывает деформацию сгибатетельных поверхностей, констрикции, аутоампутацию и облитерацию пальцев.
Характерны периорифициальные кератозы, а к сочетанным симптомам относятся разнообразные проявления дистрофии ногтей и гипотрихоза. Заболевание обычно проявляется в первые 6 месяцев жизни и прогрессирует. У ряда пациентов на пораженной коже развивается плоскоклеточная карцинома. Генетическая причина неизвестна. Большинство случаев спорадические, но могут быть и аутосомно-доминантные варианты. У детей заболевание следует отличать от рецессивных трансгредиентных ЛПК, в том числе от болезни острова Меледы, а также, ввиду присутствия периорифициальных кератозов, от энтеропатического акродерматита.
г) Другие эктодермальные синдромы с кератодермией. Ладонно-подошвенная кератодермия (ЛПК) отмечается при синдроме Негели-Франческетти-Ядассона (М1М 161000) и ретикулярной пигментной дерматопатии (MIM 125595), аллельных синдромах вследствие доминантных мутаций в неспиральных доменах E1/V1 кератина 14. К другим признакам относятся отсутствие дерматоглифики, сетчатые пигментные аномалии и другие, слабо выраженные эктодермальные дефекты. ЛПК наблюдается также в случае дефектов в кератинах 5 и 14 при тяжелой форме простого буллезного эпидермолиза Доулинга-Меара, при синдроме Киндлера вследствие мутаций в FERMT1, кодирующем киндлин-1, молекулу клеточной адгезии, участвующую в сигнализации через филаментную сеть актина, а также при эктодермальной дисплазии с хрупкостью кожи вследствие дефектов в десмосомальном белке плакофилин I.
Ладонно-подошвеннкая кератодермия (ЛПК) выявляется при гидротической эктодермальной дисплазии (синдроме Клоустона, MIM 129500) вследствие мутаций в GJB6, кодирующем коннексин 30 (Сх30), которая может имитировать врожденную пахионихию, а также при окуло-денто-дигитальной дисплазии (MIM 164200) вследствие мутаций в GJA1, кодирующем Сх43. Аналогично, ЛПК может быть частью картины вариабельной эритрокератодермии (MIM 133200) вследствие мутаций в GJB3 или GJB4.
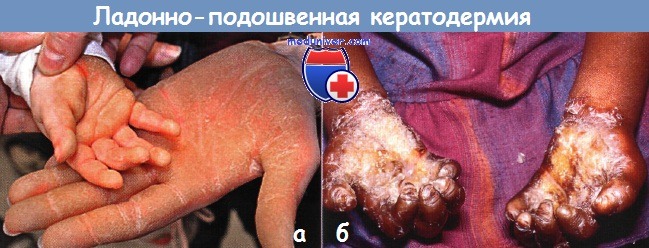
б - синдром Олмстеда.
Тяжелая рубцующая кератодермия ладоней и подошв у этой 5-летней девочки сочеталась с периоральным и перигенитальным гиперкератозом, гипергидрозом, рецидивирующими кожными инфекциями и отставанием в росте.
Системное лечение ретиноидами привело к частичному уменьшению очагов на ладонях и стопах.
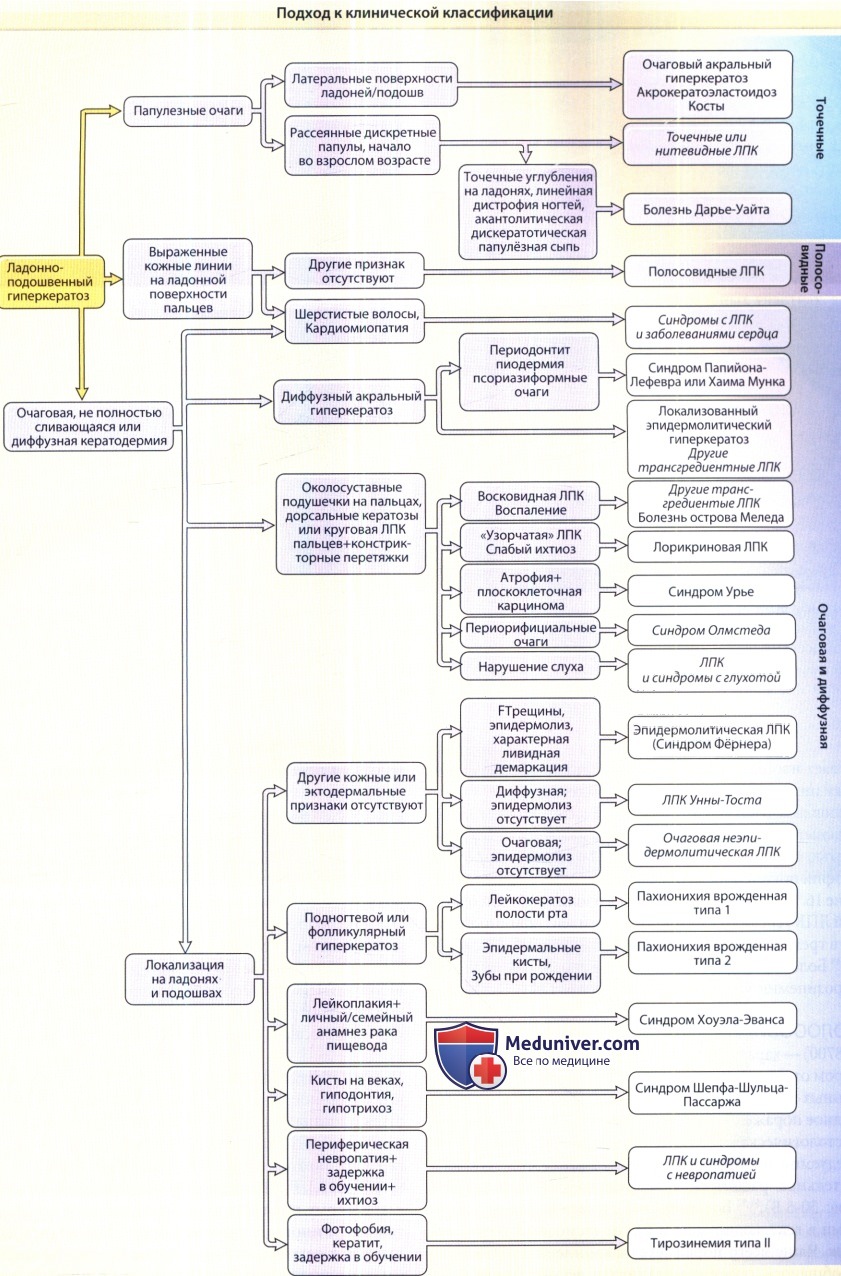
Диагнозы, выделенные курсивом, установлены прагматически, и не являются генетически обусловленными синдромами.
Тип кератодермии (например, папулезный, очаговый, полосовидный, диффузный) не следует использовать как основной параметр классификации,
поскольку эти типы могут быть разными даже у членов одной и той же семьи.
Во всех новых случаях целесообразно искать эктодермальные и синдромные ассоциации.
- Рекомендуем далее ознакомиться со статьей "Симптомы и клиника кератодермии с поражением слизистых"
Оглавление темы "Ладонно-подошвенные кератодермии (ЛПК).":- Причины и механизмы развития ладонно-подошвенной кератодермии (ЛПК)
- Клиническая классификация ладонно-подошвенной кератодермии (ЛПК)
- Симптомы и клиника кератодермии с эктодермальной дистрофией
- Симптомы и клиника кератодермии с поражением слизистых
- Симптомы и клиника кератодермии с поражением сердца
- Симптомы и клиника кератодермии с нарушением слуха
- Симптомы, клиника кератодермии с невропатией и задержкой речевого развития
- Анализы и биопсия при кератодермии
- Дифференциальная диагностика кератодермии
- Современное лечение кератодермии