
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Эритропоэтическая протопорфирия (ЭПП) - эпидемиология, этиология, клиника
Эритропоэтическая протопорфирия - краткий обзор:
- Вторая по частоте встречаемости среди кожных порфирий.
- Аутосомно доминантное или аутосомно рецессивное заболевание с недостаточностью феррохелатазы или Х-сцепленное доминантное заболевание с гиперфункцией синтазы δ-аминолевуленовой кислоты II типа.
- Начало заболевания — в раннем детском возрасте (от 1 года до 4 лет); редко может встречаться позднее начало.
- К кожным симптомам относятся эритема, отек, пурпура, утолщение кожи и восковидные рубцы; образование пузырей встречается редко.
- К симптомам также относятся острое жжение и покалывание кожи, которые могут быть очень выраженными.
- Почти у 5% пациентов может развиться фатальная печеночная недостаточность.

а) Эпидемиология. Хотя точный показатель заболеваемости эритропоэтической протопорфирией (ЭПП) неизвестен, частота сообщений о заболевании в популяции с момента открытия болезни в 1961 году нарастает, таким образом можно заключить, что это вторая по частоте встречаемости форма заболевания после ППК. В мире заболевание возникает с частотой от 1:75000 до 1:200000.
Последний обзор демографических, клинических, биохимических и генотипичских характеристик эритропоэтической протопорфирии в Швеции показал, что распространенность заболевания составляет 1:180000, а самым частым возрастом манифестации заболевания является первый год жизни, тогда как средний возраст, когда впервые устанавливается диагноз, приходится на 22 года.
б) Этиология эритропоэтической протопорфирии (ЭПП). Специфическим дефектным ферментом при ЭПП является феррохелатаза, которая у пораженных пациентов снижена в различных клетках, включая эритроциты, митоген-стимулированные лимфоциты и культивированные фибробласты кожи у пациентов, страдающих этим заболеванием, а также у носителей гена. Феррохелатаза находится на матриксной поверхности внутренней митохондриальной мембраны и катализирует финальную стадию образования гема, введение двухвалентного железа (Fe2+) в ПРТО.
Этот богатый лизином фермент имеет молекулярную массу 63000 а.е.м. и оптимальный уровень pH 7,8, и также называется гем-синтазой или прото-гем-ферролиазой. В отличие от других ферментов в процессе биосинтеза гема, который в качестве субстрата требует редуцированные формы порфиринов (порфириногены), феррохелатаз использует ПРОТО — окисленный изомер протогена. Конечный продукт этого процесса — феррат ПРОТО или гем, диффундирует из митохондрий в цитоплазму, где становится доступным в качестве протетической группы после комбинирования с соответствующими апопротеинами.
В зависимости от уровня потребления гема в цитоплазме, гем контролирует свой собственный синтез, основываясь на потребностях клетки.
Была клонирована и секвенирована кДНК для феррохелатазы человека, и было показано, что она имеет 88% гомологии с ферментом мыши. мРНК феррохелатазы человека кодируется структурным ядерным геном, имеющим такое же название, ген феррохелатазы (FECH), который расположен на хромосоме 18q21.3 и насчитывает почти 45 кб геномной ДНК. Ген состоит из 11 экзонов и 10 интронов. В молекулярно-генетических исследованих у пациентов с ЭПП было выявлено более 60 мутаций разных типов, при этом наиболее частыми явились делеции и инсертационные мутации.
Молекулярная эпидемиология эритропоэтической протопорфирии включает три паттерна наследования. В большинстве случаев, болезнь наследуется по псевдоаутосомнодоминантному типу, т.е., проявления болезни возникают только при сочетании с мутацией феррохелатазы, которая наследуется посредством транс-сплайсинга гипоморфной аллели (FECHIVS3-48C,c.315-48C). Эта добавочная нуклеотидная перестройка ведет к модуляции сплайсинга и постоянному использованию искаженного акцепторного сайта сплайсинга. Неправильно сплайсированная мРНК разрушается путем антисмыслового распада, при чем образуется мРНК с пониженным уровнем стабильности. В результате наблюдается дополнительное снижение резидуальной активности феррохелатазы примерно на 15-40%, что сокращает каталитическую активность вплоть до клинических проявлений эритропоэтической протопорфирии.
Второй паттерн наследования эритропоэтической протопорфирии (около 5%) аутосомно рецессивный, при котором мутации феррохелатазы возникают в обеих аллелях, и третий паттерн включает в себя С-концевую делецию в гене синтазы б-аминолевуленовой кислоты II типа, что вызывает патологическое функционирование белка, и как следствие эритропоэтическую протопорфирию без анемии и перенасыщения железом. Это и есть Х-сцепленное доминантное наследование.
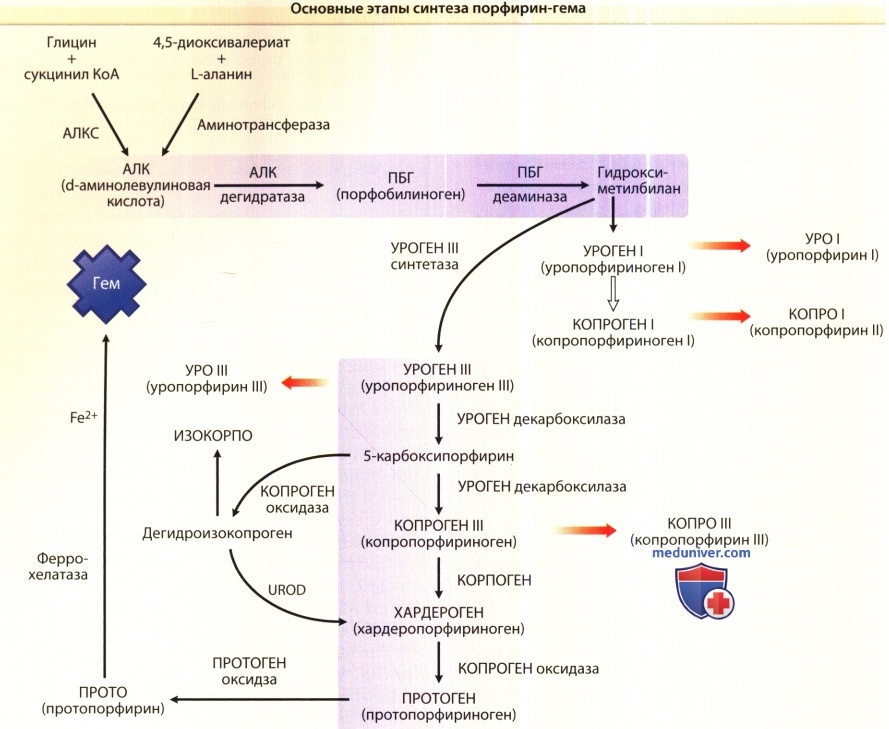
Аминолевуленовая кислота может образовываться из глицина и сукцинилКоА(СКоА), что является основным источником у млекопитающих и катализируется митохондриальным энзимом АЛА синтазой (АЛАС).
Две молекулы АЛА формируют монопирроль порфобилиногена (ПБГ) в реакции, катализированной ферментом АЛА дегидратазой (АЛАД).
Четыре молекулы ПБГ упакованы с помощью ПБГ деаминазы (ПБГД), также известной как гидроксиметилбилансинтаза (ГМБС), в линейный тетрапиррол, гидрокисметилбилан (ГМБ), который может спонтанно зациклироваться и образовывать уропорфириноген (УРОГЕН) 1.
Четыре ацетильные группы УРОГЕНа 1 последовательно декарбоксилируются уропорфириногендекарбоксилазой (УРОД), чтобы образовать копропорфириноген (КОПРОГЕН) 1.
ГМБ также может превращаться в УРОГЕН III с помощью фермента уропрфириноген III синтазы (УРОС), в этой реакии одно из колец монопирроля «опрокидывается», что меняет последовательность концевых цепей.
Ацетильная группы УРОГЕН III последовательно декарбоксилируются с помощью УРОД до КОПРОГЕН III.
КОПРОГЕН III превращается в протопорфириноген (ПРОТОГЕН) IX с помощью фермента копропорфириноген оксидазы (КПОКС), которая оксидативно декарбоксилирует каждую пропионильную группу.
ПРОТОГЕН IX превращается в протопорфирин (ПРОТО) IX с помощью протопорфириногеноксидазы (ППОКС). ПРОТО IX превращается в гем с помощью феррохелатазы (ФЕХ), которая катализирует внедрение в молекулу закис-ных соединений железа.
в) Клиника эритропоэтической протопорфирии (ЭПП):
1. Кожа. Обычно заболевание начинается в раннем возрасте и характеризуется острыми эпизодами кожной фоточувствительности, включая чувство жжения и покалывания (жгучей боли) и зуд, поражающий кожу, подверженную солнечному излучению, в особенности на носу, щеках и тыльных поверхностях кистей. Эти симптомы сопровождаются эритемой, отеком, угревой сыпью и, реже, пурпурой. Симптомы могут возникать уже после нескольких минут, проведенных на солнце, они, как правило, носят сезонный характер, начинаясь ранней весной, наблюдаются на протяжении всего лета и постепенно стихают к зиме.
Кожные поражения часто разрешаются медленно, оставляя небольшие атрофичные восковидные или изрытые рубцы. Может отмечаться сморщивание кожи вокруг рта (псевдотрещины). Кожа пальцев, особенно пястно-фаланговых и межфаланговых суставов, часто выглядит утолщенной, покрытой морщинами и восковидной, что наводит на мысль о преждевременном старении (так называемые старческие костяшки у ребенка). Этот коварный симптом является патогномоничным. На лице возникают поверхностное рубцевание спинки носа и небольшие кольцевидные неглубокие рубцы. При умеренном климате везикулярные и буллезные поражения встречаются редко, получены данные об их возникновении у пациентов, подверженных облучению тропическим солнцем. У большинства больных поражение кожи персистирует на протяжении всей жизни, хотя у некоторых пациентов, со временем симптомы могут становиться менее выраженными.
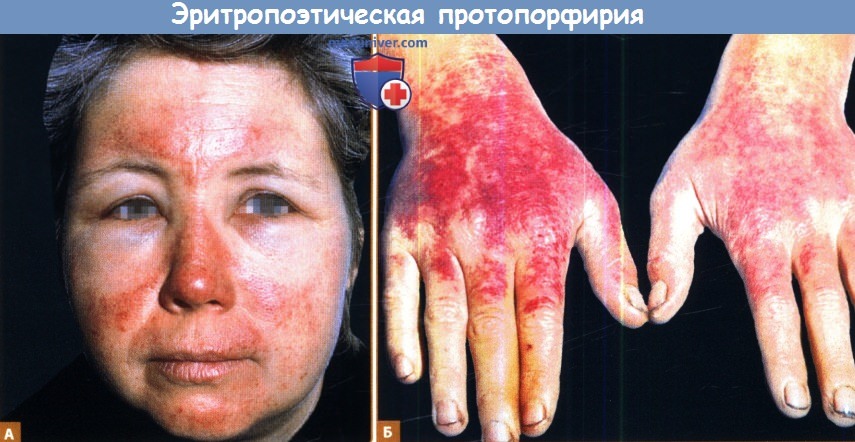
А. Через час после солнечного воздействия, лицо 35-летней женщины отечное с покраснениями, также на области лба, щек и носа определяется пурпура.
Пациентка испытывает сильную жгучую боль в области поражения.
Б. Отек и пурпура на дорсальной поверхности ладоней после солнечного воздействия.
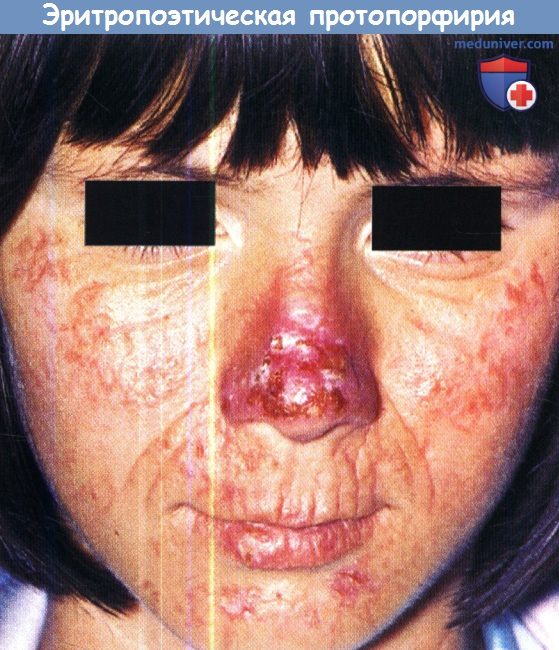
15-летняя пациентка с эритропоэтической протопорфирией с тяжелой светочувствительностью.
На коже носа, нижней губе и подбородке отмечаются эритематозные, местами эрозивные, и покрытые корками элементы.
На носу и губах видны эритематозные поражения, несколько небольших слегка вдавленных рубцов и специфическое восковидное утолщение кожи.
Между носом и ртом, а также на щеках и лице отмечаются линейные рубцы.
2. Печень. У пациентов с ЭПП иногда возникает тяжелая, угрожающая жизни гепатотоксичность. Протопорфирин — это липофильная молекула, метаболизм которой осуществляется в печени, в связи с чем, эти пациенты имеют высокий риск развития холелитиаза и холестаза, что в конечном счете ухудшает функцию печени. Это может усугубляться гемолизом эритроцитов, нагруженных ПРОТО, что создает порочный круг повышенного эритропоэза и повышенной нагрузки ПРОТО на печень. Конечным итогом таких изменений является терминальная печеночная недостаточность, развивающаяся почти у 5% пациентов с ЭПП. У больных развивается тяжелая желтуха, цирроз печени, они могут впасть в печеночную кому и умереть. У некоторых пациентов с ЭПП и терминальной печеночной недостаточностью может развиваться аксональная нейропатия, напоминающая таковую при печеночных порфириях, за исключением нормальных АЛК и ПБГ. Факторы риска развития ЭПП-ассоциированной печеночной недостаточности остаются неизвестными, но вероятным объяснением является паренхматозная кристаллизация не полностью очищенного печеночного ПРОТО.
Митоферрин-1 (МФР1) — первый из двух типов гомологичных транспортеров железа в митохондриях, который отвечает за митохондриальный транспорт железа в развивающихся клетках эритропоэтического ряда. Делеция МФР1 в гепатоцитах не имеет клинических проявлений или биохимических последствий в нормальных условиях, однако при повышенном синтезе порфирина, такая делеция в гепатоцитах ведет к снижению способности конвертировать протопорфирин IX в гем, что вызывает протопорфирию, холестаз и мосто-видный фиброз печени. Нарушения синтеза гема могут иметь гепатотоксический эффект.139 Экспериментальные исследования показали, что перфузия ПРОТО в печени крыс вызывает доза-зависимый холестаз, который может усиливать гепатотоксичность ПРОТО. Печеночная недостаточность также развилась у двух сибсов, унаследовавших ЭПП по аутосомно-рецессивному типу, как компаунд-гетерозиготы, что позволяет предположить, что наличие такого генотипа может каким-то образом усиливать риск данного осложнения. Однако это является спорным вопросом, так как у некоторых носителей компаунд-гетерозиготы сохраняется нормальная печеночная функция, в то время как у других развивается печеночная недостаточность.
Кожные и печеночные симптомы являются основными признаками эритропоэтической протопорфирии. При данном заболевании не отмечается гипертрихоза, мили или гиперпигментации, как это бывает при поздней порфирии кожи (ППК), а также эритродонтоза, как при ВЭП. Гемолитическая анемия является абсолютно нетипичной,хотя почтиу 11% пациентов с ЭПП отмечается легкая анемия неясной этиологии. Получены данные о холецистолитиазе у некоторых пациентов с ЭПП в относительно раннем возрасте; в одном исследовании у 12% пациентов был выявлен холестаз, трем пациентам была выполнена холецистэктомия.
- Рекомендуем далее ознакомиться со статьей "Анализы и гистология при эритропоэтической протопорфирии (ЭПП)"
Редактор: Искандер Милевски. Дата публикации: 30.11.2018