
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) поликистоза почек взрослых
Поликистозная болезнь почек с аутосомно-доминантным типом наследования с высокой пенетрантностью (взрослый тип) — наследственное заболевание, которое характеризуется множественными кистами в обеих почках, разрушающими паренхиму почки, что приводит к почечной недостаточности. Это заболевание является причиной 5-10% случаев хронической почечной недостаточности с необходимостью диализа и последующей трансплантации почки. Частота заболевания составляет 1 случай на 400-1000 живорожденных детей.
Несмотря на доминантный тип наследования, для формирования клинической картины необходимы мутации в обоих аллелях гена PKD. Заболевание всегда двухстороннее. Кисты первоначально вовлекают только часть нефронов, поэтому функция почек сохраняется до 30-50 лет. Поликистозная болезнь почек (взрослый тип) — генетически гетерогенное заболевание.
Исследования семей позволили установить, что гены PKD располагаются на хромосомах 16р13.3 (PKD1) и 4q21 (PKD2), редко выявляются мутации по крайней мере одного дополнительного гена, ассоциированного с заболеванием. Мутации PKD1 имеют 85% пациентов (большинство остальных имеют мутации PKD2). Мутации PKD1 ассоциируются с более тяжелым течением заболевания, развитием терминальной стадии хронической болезни почек и ранней смертью (53 года при мутациях PKD1 vs 69 лет при мутациях PKD2).
При мутации PKD1 вероятность развития почечной недостаточности до 40 лет составляет менее 5%, до 50 лет — 35%, к 60 годам — 70% и к 70 годам превышает 95%. При мутации PKD2 вероятность развития почечной недостаточности до 50 лет составляет менее 5%, к 60 годам — ~ 15%, после 70 лет — 45%. Несмотря на преимущественное поражение почек, поликистозная болезнь почек с аутосомно-доминантным типом наследования — системное заболевание, при котором кисты и другие аномалии наблюдаются и в других органах.
а) Генетика и патогенез. Широкий спектр мутаций PKD1 и PKD2 (гетерогенность аллелей) усложняет генетическую диагностику заболевания.
Ген PKD1 кодирует большой интегральный белок полицистин-1 (молекулярная масса 460 кДа), который имеет крупную внеклеточную область, множественные трансмембранные домены и короткий цитоплазматический хвост. Белок располагается в клетках эпителия канальцев в основном дистального отдела нефрона. Точная функция белка еще неизвестна, но установлено, что полицистин-1 содержит домены, которые участвуют во взаимодействии клеток между собой и с матриксом.
Ген PKD2 кодирует интегральный мембранный белок полицистин-2. Этот белок располагается во всех сегментах канальцев почек, а также экспрессируется во многих экстраренальных тканях. Полицистин-2 функционирует как ионный канал, проницаемый для Са2+, а важнейшим дефектом при поликистозной болезни почек (взрослый тип) является нарушение регуляции уровня Са2+ внутри клеток.
Патогенез поликистозной болезни почек до конца не ясен, но предполагают, что в основе патологии лежит повреждение реснично-центросомного комплекса эпителия канальцев. Клетки почечного эпителия канальцев имеют одиночные неподвижные первичные реснички (нитеподобные органеллы длиной 2-3 мкм), которые находятся на апикальной поверхности этих клеток и выступают в просвет канальца. Реснички состоят из микротрубочек, прикрепленных к базальному телу центриоли.
Реснички являются частью системы органелл и клеточных структур, чувствительных к механическим сигналам (механосенсоров). Считается, что реснички улавливают изменения направления движения и давления жидкости в канальцах, а межклеточные контактные комплексы контролируют силу сцепления клеток между собой и с матриксом. В ответ на внешние раздражители механосенсоры могут регулировать поток ионов (реснички могут индуцировать поток Са2+ в культуре эпителиальных клеток почек), полярность и пролиферацию клеток.
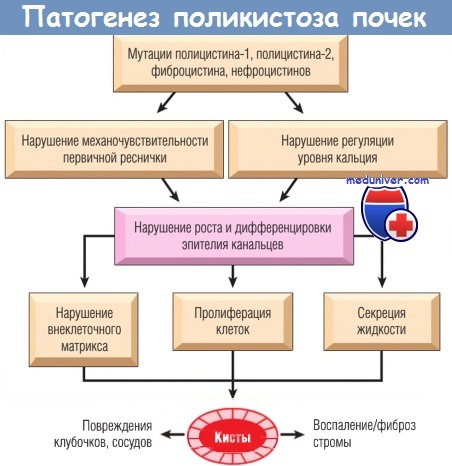
Предположение, что в основе формирования кист лежат дефекты механосенсоров, потока Са2+ и проведения сигнала, подтверждается результатами некоторых исследований:
- полицистин-1 и полицистин-2 располагаются в первичной ресничке. Мутации других генов при кистозной болезни почек (например, мутации гена NPHP) также затрагивают синтез белков, расположенных в ресничках и/или базальных тельцах;
- в нокаутных по гену PKD1 организмах (червь С. elegans) отмечались аномалии ресничек и формирование кист [92];
- в клетках эпителия канальцев у мышей с делецией гена PKD1 (обусловливающей летальность эмбрионов у этих мышей) реснички имели нормальную структуру, но в них отсутствовал поток Са2+, индуцируемый током жидкости, который наблюдается в нормальных клетках канальцев.
Полицистин-1 и полицистин-2 могут формировать белковый комплекс, участвующий в регуляции внутриклеточного Са2+ в ответ на ток жидкости. Вероятно, поток жидкости в почечных канальцах приводит к изгибу реснички, что открывает кальциевые каналы. Результатом мутации любого гена PKD может стать потеря полицистинового комплекса или формирование аберрантного комплекса. Последующее нарушение нормальной активности полицистина приводит к изменению уровня внутриклеточного Са2+. Это влияет на пролиферацию клеток, базальный уровень апоптоза, взаимодействие с ВКМ и секреторную функцию эпителия и приводит к развитию характерных симптомов поликистозной болезни почек (взрослый тип).
Взаимодействие продуктов генов PKD1 и PKD2, вероятно, обусловливает один и тот же фенотип болезни, вызываемой мутацией любого из двух этих генов. Увеличение количества клеток в результате нарушения пролиферации и повышение объема жидкости в просвете канальцев из-за гиперсекреции эпителия, выстилающего кисту, приводят к ее прогрессирующему росту. Кроме того, в кисте накапливаются медиаторы, выделяемые эпителиальными клетками, которые усиливают секрецию жидкости и индуцируют воспаление. Все это приводит к дальнейшему увеличению кист и интерстициальному фиброзу, характерному для прогрессирующей поликистозной болезни почек.
б) Морфология. При макроскопическом исследовании обе почки значительно увеличены (масса одной почки может достигать 4 кг). Внешняя поверхность почек представлена только кистами, без нормальной паренхимы. Диаметр кист — В-4 см. При микроскопическом исследовании между кистами выявляют функционирующие нефроны. Кисты могут быть заполнены прозрачной серозной или, чаще, мутной красно-коричневой жидкостью, иногда геморрагической. Кисты формируются из разных отделов нефрона, поэтому выстланы различным эпителием. Иногда формируются папиллярные эпителиальные структуры и полипы, выступающие в просвет кисты.
Изредка в формирование кист вовлекаются капсулы Боумена, при этом в просвете кисты выявляются элементы клубочка.
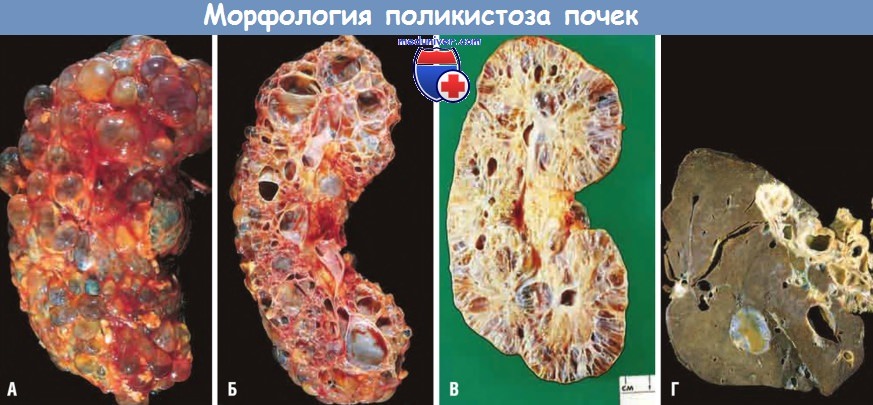
внешний вид почки (А) и на разрезе (Б). Почки значительно увеличены, содержат множественные кисты.
(В) Поликистозная болезнь почек с аутосомно-рецессивным типом наследования (детский тип). Меньшее нарушение структуры почки.
(Г) Кисты в печени при поликистозной болезни почек (взрослый тип).
в) Клинические признаки. До развития почечной недостаточности заболевание может протекать бессимптомно. Иногда кровоизлияние или прогрессирующее растяжение кист сопровождается болью, а выделение сгустков крови — почечной коликой. При пальпации живота выявляются увеличенные почки, при этом могут возникать тянущие боли. Изредка заболевание манифестирует внезапной гематурией и другими признаками прогрессирующей почечной недостаточности: протеинурией (редко более 2 г/сут), полиурией, гипертензией. У пациентов с мутациями гена PKD2 клинические симптомы появляются в более старшем возрасте, чем при мутациях гена PKD1, и позже развивается почечная недостаточность.
На тяжесть течения болезни влияют как генетические факторы, так и факторы окружающей среды. Почечная недостаточность быстрее прогрессирует у лиц с темным цветом кожи (тесно коррелирует с признаками серповидноклеточной анемии) и у мужчин с гипертензией.
У пациентов с поликистозной болезнью почек обнаруживают и другие врожденные экстраренальные аномалии. Около 40% пациентов имеют в печени одну или несколько кист из эпителия желчных протоков (поликистозная болезнь печени), обычно это заболевание бессимптомно. Значительно реже встречаются кисты селезенки, поджелудочной железы и легких. Мешковидные аневризмы сосудов виллизиева круга (возможно, из-за нарушений экспрессии полицистина в гладкомышечных клетках стенки артерий) и субарахноидальные кровоизлияния, обусловленные этими аневризмами, приводят к летальному исходу в 4-10% случаев.
Пролапс митрального клапана и другие аномалии клапанов сердца встречаются у 20-25% пациентов, но большинство из этих аномалий бессимптомны. Клинический диагноз ставят с помощью методов радиологической визуализации.
Хроническая почечная недостаточность на фоне поликистозной болезни почек (взрослый тип) отмечается у пациентов, которые в течение многих лет живут с азотемией, медленно прогрессирующей до уремии. В итоге 40% взрослых пациентов умирают от коронарной и гипертонической болезней, 25% — от инфекции, 15% — от разрыва мешковидной аневризмы сосудов вилизиева круга или кровоизлияния в мозг на фоне гипертонической болезни, остальные — от других причин.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) поликистоза почек детей"
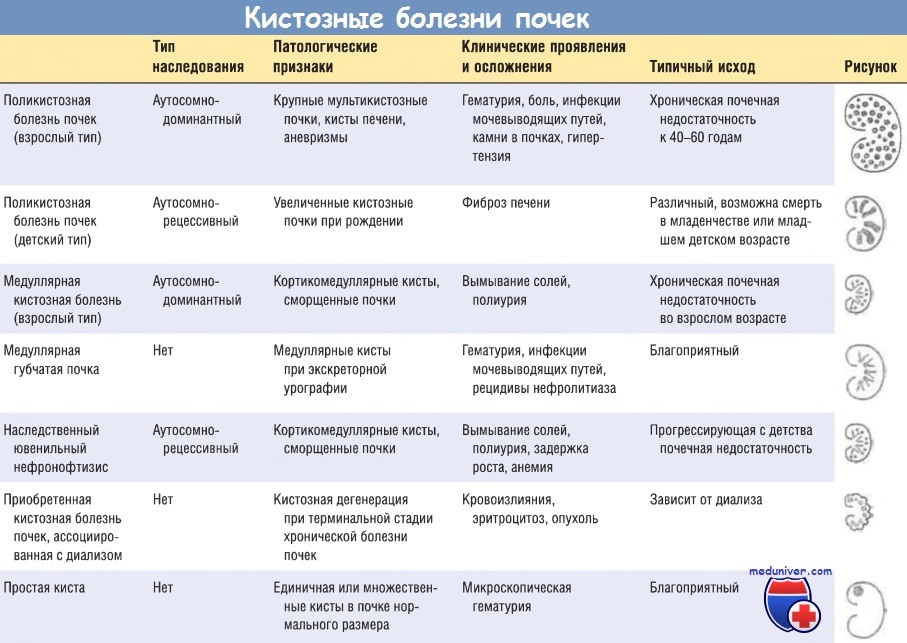
Оглавление темы "Болезни почек":- Механизм развития (патогенез) мультикистозной дисплазии почки
- Механизм развития (патогенез) поликистоза почек взрослых
- Механизм развития (патогенез) поликистоза почек детей
- Механизм развития (патогенез) медуллярной болезни почек
- Механизм развития (патогенез) кист почек от гемодиализа
- Механизм развития (патогенез) простой кисты в почке
- Механизм развития (патогенез) гидронефроза
- Механизм развития (патогенез) камней в почках
- Механизм развития (патогенез) доброкачественных опухолей почек
- Механизм развития (патогенез) рака почки - аденокарциномы