
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) гемофилии А
Гемофилия А (дефицит фактора VIII) — наиболее частое наследственное заболевание, характеризующееся опасными для жизни кровотечениями. Гемофилия А обусловлена мутациями фактора VIII, выполняющего важную функцию кофактора фактора IX в каскаде коагуляции.
Гемофилия А ассоциируется со сцепленным с Х-хромосомой рецессивным наследованием, поэтому главным образом поражает лиц мужского пола и гомозиготных женщин.
В редких случаях чрезмерное кровотечение возможно у гетерозиготных женщин, предположительно в результате случайной инактивации Х-хромосомы, несущей нормальный аллель фактора VIII, в большинстве клеток (неблагоприятная лайонизация).
У 30% пациентов заболевание в семейном анамнезе отсутствует, причиной служат новые мутации.
При гемофилии А тяжесть клинических проявлений значительно варьирует в зависимости от активности фактора VIII.
Если она составляет менее 1% нормы, заболевание протекает тяжело; при уровне 2-5% наблюдают умеренное расстройство, а у лиц с 6-50% нормы заболевание протекает легко. Различная степень дефицита фактора VIII в значительной мере определяется гетерогенностью соответствующих мутаций. Как и в случае b-талассемии, генетическими нарушениями могут быть делеции, нонсенс-мутации, создающие стоп-кодоны, и мутации, вызывающие ошибки сплайсинга мРНК.
Наиболее тяжелый дефицит наблюдается в случае инверсии, затрагивающей Х-хромосому, когда синтез фактора VIII полностью блокирован. Реже тяжелая гемофилия А ассоциируется с точечными мутациями гена фактора VIII, нарушающими функцию белка. В таких случаях уровень фактора VIII оказывается нормальным при определении иммунологическими методами.
Мутации, при которых синтезируется фактор VIII с некоторой степенью активности, приводят к развитию заболевания от легкой до умеренной степени. У таких пациентов расстройство может быть модифицировано другими генетическими факторами, влияющими на уровень экспрессии фактора VIII, который широко варьирует у здоровых людей.
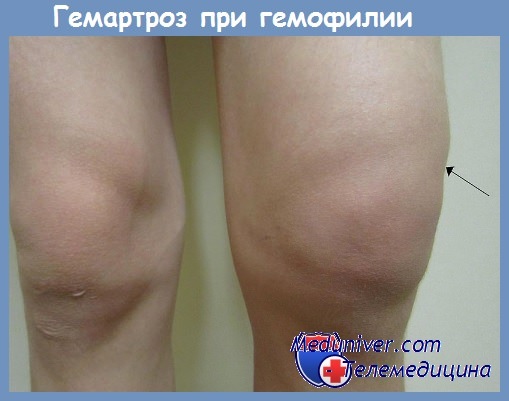
При гемофилии А отмечается тенденция к легкому возникновению кровоподтеков и массивных геморрагий после травм и хирургических вмешательств. Кроме того, часто происходят спонтанные геморрагии в тех участках тела, которые в норме подвержены травмам, особенно в суставах. Повторные кровотечения в суставы приводят к прогрессирующей их деформации, которая может стать необратимой. Характерно отсутствие петехий.
Пациенты с гемофилией А, как правило, имеют увеличенное частичное тромбопластиновое время и нормальное протромбиновое время, что свидетельствует о нарушении внутреннего пути коагуляции.
Для диагностики необходимо использовать тесты, специфичные для фактора VIII.
Почему у пациента с гемофилией А возникают кровотечения, хотя внешний путь коагуляции не нарушен? Очевидно, коагуляция in vitro представляет собой несовершенную модель того, что происходит in vivo.
Вероятно, при дефиците фактора VIII отложение фибрина не в состоянии обеспечить гемостаз (детальное обсуждение этой проблемы не входит в нашу задачу). Вероятно, основная роль внешнего пути коагуляции в гемостазе заключается в инициации ограниченной активации тромбина в случае повреждения тканей. Этот первоначальный прокоагулянтный стимул усиливается и амплифицируется посредством обратной связи, благодаря которой тромбин активирует факторы XI и IX внутреннего пути коагуляции.
В отсутствие фактора VIII эта обратная связь неактивна, и образуется недостаточно тромбина (и фибрина), чтобы обеспечить формирование стабильного сгустка крови.
Факторы свертывания крови
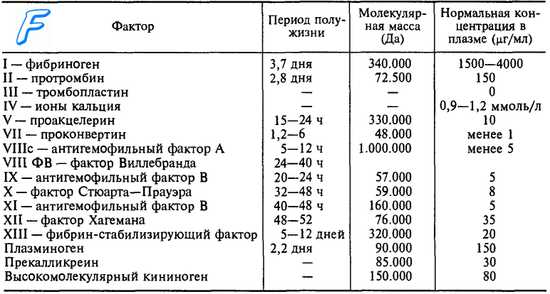
Кроме того, высокий уровень тромбина необходим для активации ингибитора фибринолиза, активируемого тромбином. Таким образом, неадекватная коагуляция (неадекватный фибриногенез) и неадекватное удаление сгустка (неадекватный фибринолиз) способствуют геморрагиям при гемофилии.
На сегодняшний день трудно объяснить существующую у больных гемофилией тенденцию к геморрагиям определенной локализации (суставы, мышцы и ткани центральной нервной системы).
Гемофилию А лечат введением рекомбинантного фактора VIII. У 15% пациентов с тяжелой гемофилией А образуются антитела, связывающие и ингибирующие фактор VIII.
Видимо, этот белок не распознается иммунной системой и воспринимается как чужеродный, что существенно снижает терапевтический эффект.
До использования рекомбинантного фактора VIII больные гемофилией, получая приготовленный из плазмы доноров концентрат фактора VIII, имели высокий риск заражения ВИЧ и тысячи больных гемофилией заболевали СПИДом.
Позднее риск передачи ВИЧ был устранен, однако слишком поздно для целого поколения больных гемофилией. Попытки разработать методы соматической генотерапии при гемофилии продолжаются.

- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) гемофилии В"
Оглавление темы "Патогенез болезней":- Механизмы развития (патогенез) тромботической микроангиопатии
- Механизмы развития (патогенез) кровотечений при нарушении функции тромбоцитов
- Механизмы развития (патогенез) кровотечений при дефиците факторов коагуляции
- Механизмы развития (патогенез) болезни Виллебранда
- Механизмы развития (патогенез) гемофилии А
- Механизмы развития (патогенез) гемофилии В
- Механизмы развития (патогенез) ДВС
- Строение и функция легких
- Виды врожденных аномалий легких
- Механизмы развития (патогенез) ателектаза легкого