
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) болезни Виллебранда
Болезнь Виллебранда (дефицит WF) — наиболее частое наследственное геморрагическое заболевание (в США им болеет 1% взрослого населения).
У большинства пациентов склонность к геморрагиям обычно выявляют случайно, например при хирургическом или стоматологическом вмешательстве или других видах гемостатического стресса.
Характерные симптомы — спонтанные кровотечения из слизистых оболочек (например, эпистаксис), массивное кровотечение из ран, меноррагия и удлинение времени кровотечения при нормальном количестве тромбоцитов. Заболевание наследуется, как правило, по аутосомно-доминантному типу.
На молекулярном уровне болезнь Виллебранда гетерогенна. Описано более 20 вариантов, которые можно разделить на две большие категории.

Болезнь Виллебранда типа 1 (аутосомно-доминантный тип наследования) и болезнь Виллебранда типа 3 (аутосомно-рецессивный тип наследования) ассоциируются со сниженным количеством циркулирующего WF.
Тип 1 характеризуется дефицитом WF (от слабой до умеренной степени) и составляет 70% всех случаев. Как правило, присутствуют неполная пенетрантность и вариабельная экспрессивность, однако в целом тип 1 является легкой формой заболевания.
Тип 3 характеризуется тяжелым дефицитом WF и, соответственно, более серьезными клиническими проявлениями. Поскольку выраженный дефицит WF оказывает заметное влияние на стабильность фактора VIII, некоторые геморрагии напоминают таковые при гемофилии. Природа мутаций у большинства пациентов с болезнью Виллебранда типа 1 определена плохо. В некоторых случаях обнаружены миссенс-мутации. Причиной заболевания типа 3 служат делеции или мутации рамки считывания обоих аллелей.
Для болезни Виллебранда типа 2 характерны качественные дефекты WF. Существует несколько подтипов болезни Виллебранда типа 2, наиболее распространен тип 2А (аутосомно-доминантный тип наследования). WF экспрессируется в нормальном количестве, однако присутствуют миссенс-мутации, приводящие к дефекту сборки мультимеров. Крупные и промежуточной величины мультимеры, наиболее активные формы WF, исчезают из плазмы. Болезнь Виллебранда типа 2 составляет 25% всех случаев заболевания и ассоциируется с геморрагией слабой или умеренной степени.
У пациентов с болезнью Виллебранда функция тромбоцитов, несмотря на их нормальное количество, нарушена. Уровень активного WF в плазме, определяемый в тесте с ристоцетином, снижен. Поскольку WF стабилизирует фактор VIII, дефицит WF приводит к вторичному снижению уровня фактора VIII. Это отражается в удлинении частичного тромбопластинового времени при болезни Виллебранда типов 1 и 3. Однако осложнения, типичные для тяжелого дефицита фактора VIII, например кровоизлияния в суставы, наблюдаются только у немногих пациентов с болезнью Виллебранда типа 3.
Таким образом, наследственные дефекты WF при болезни Виллебранда приводят к вторичным нарушениям адгезии тромбоцитов и образованию сгустка. Даже в семьях, в которых сегрегируется один дефектный аллель WF, обычна широкая вариабельность клинической экспрессии.
Частично это обусловлено дополнительными генетическими факторами, влияющими на уровень циркулирующего WF и существенно варьирующими в норме. Для лечения лиц с нарушениями гемостаза можно использовать десмопрессин, стимулирующий высвобождение WF, или концентраты плазмы, содержащие фактор VIII и WF.

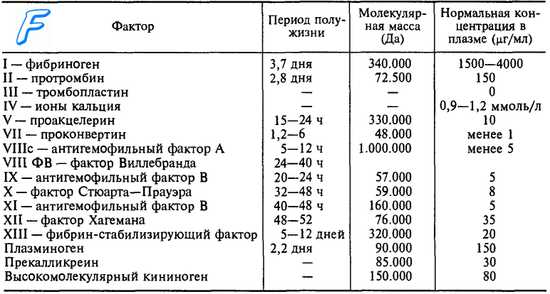
Факторы свертывания крови
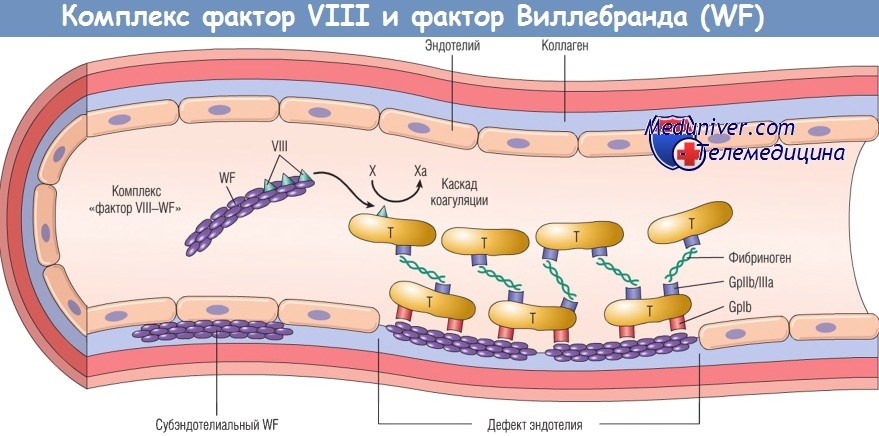
Фактор VIII синтезируется в печени и почках, фактор Виллебранда (WF) — в эндотелиальных клетках и мегакариоцитах.
Оба фактора связываются, образуя комплекс. WF присутствует также в субэндотелиальном матриксе нормальных кровеносных сосудов и а-гранулах тромбоцитов (Т).
После повреждения эндотелия экспонирование субэндотелиального WF вызывает адгезию тромбоцитов, в основном посредством рецептора тромбоцитов гликопротеина Ib (GpIb).
Циркулирующий WF и WF, высвобождаемый из а-гранул активированных тромбоцитов, способны связываться с экспонированным субэндотелиальным матриксом,
еще более усиливая адгезию и активацию тромбоцитов.
Активированные тромбоциты образуют гемостатические агрегаты; фибриноген (а возможно, и WF) участвует в агрегации,
взаимодействуя с образованием мостиков с рецептором тромбоцитов гликопротеином IIb/IIIa (GpIIb/IIIa).
Фактор VIII участвует в каскаде коагуляции как кофактор в активации фактора X на поверхности активированных тромбоцитов.
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) гемофилии А"
Оглавление темы "Патогенез болезней":- Механизмы развития (патогенез) тромботической микроангиопатии
- Механизмы развития (патогенез) кровотечений при нарушении функции тромбоцитов
- Механизмы развития (патогенез) кровотечений при дефиците факторов коагуляции
- Механизмы развития (патогенез) болезни Виллебранда
- Механизмы развития (патогенез) гемофилии А
- Механизмы развития (патогенез) гемофилии В
- Механизмы развития (патогенез) ДВС
- Строение и функция легких
- Виды врожденных аномалий легких
- Механизмы развития (патогенез) ателектаза легкого