
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
История открытия галактоземии
Beuss впервые высказал предположение, что галактоземия представляет собой одну из «врожденных ошибок обмена». В 1917 году Goppert наблюдал галактозурию при «врожденном семейном поражении печени».
Выяснению метаболического блока при наследственных аномалиях обмена галактозы способствовали труды Leloir, а также Ley и Doudoroff по изучению основного и побочных путей преобразования галактозы у микроорганизмов, млекопитающих и человека.
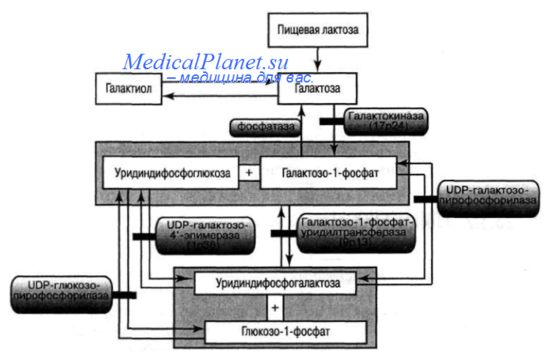
Главный путь преобразования галактозы в глюкозу известен в литературе, как путь Лелуа. Первым этапом преобразования служит фосфорилирование галактозы при участии галактокиназы (ЛТФ: галактозо-1-фосфаттрансфераза), при этом образуется га-лактозо-1-фосфат. Фосфорилирование галактозы осуществляется в печени, мозге, эритроцитах, причем реакция необратима. Дальнейшее превращение галактозо-1-фосфата обеспечивается специфическим ферментом галактозо-1-фосфат-уридилтрансферазой и требует обязательного присутствия нуклеотида — уридиндифосфатглюкозы (УДФ-глюкозы), который является кофактором этого этапа преобразования. Галактозо-1-фосфат-уридилтрансфераза имеется в клетках печени, эритроцитах, лейкоцитах и фибробластах.
Образующаяся в результате реакции УДФ-галактоза подвергается эпимеризации у 4-го углеродного атома и превращению в УДФ-глюкозу. Этот процесс происходит при участии фермента УДФ-галактозо-4-эпимеразы, который раньше назывался вальденазой.
Реакция эпимеризации обратима, и поэтому УДФ-галактоза может образовываться в организме из УДФ-глюкозы, вследствие этого галактоза пищи не является единственным ее источником для организма, а включение галактозы в нервную или соединительную ткань происходит даже при полном исключении этого сахара из пищи.
Заключительным этапом преобразования галактозы является высвобождение глюкозо-1-фосфата, который после превращения в глюкозо-6-фосфат включается в метаболические пути обмена глюкозы.
Кроме основного пути обмена галактозы (пути Лелуа), существуют дополнительные, или обходные, не играющие существенной роли в физиологических условиях, однако их следует принимать во внимание при изучении патологии обмена галактозы (преобразование галактозы через галактоповую кислоту в D-ксилулезу, образование УДФ-галактозы при участии УТФ- и УДФ-галактозо-пирофосфорилазы).
Большое значение для раскрытия патогенеза наследственных аномалий обмена галактозы имели экспериментальные исследования Kosterlitz, который установил, что в печени животных, получавших с пищей избыток галактозы, накапливается галактозо-1-фосфат. В 1955 году Schwarz и сотр. обнаружили значительные количества галактозо-1-фосфата в эритроцитах больных галактозсмией.
Kalckar и сотр. в 1956 году установили природу ферментного дефекта при наследственной галактоземии — низкую активность галактозо-1-фосфат-уридилтрансферазы в печени и эритроцитах больных. После этого внимание исследователей было направлено на разработку химических методов выявления метаболического блока, а также на изучение токсичности продуктов метаболизма галактозы и генетики галактоземии. В частности, были разработаны прямые и косвенные методы определения активности ферментов обмена галактозы, нашедшие применение в клинических исследованиях (Anderson и соавт., Schwarz, Beutler и соавт.).
В 1964 году Guthrie предложил для целей массового обследования детей микробиологический тест, a Beutler и Baluda в 1966 году разработали флюорометрический экспресс-метод, вследствие чего стало возможным проведение популяциопных генетических исследований.
Изучению клинического полиморфизма наследственных аномалий обмена галактозы способствовали исследования фенотипических вариантов галактозо-1-фосфат-уридилтрансферазы и галактокиназы с помощью метода электрофореза в крахмальном геле.
Благодаря усовершенствованию методов получения количественных и качественных характеристик ключевых ферментов обмена галактозы были выявлены различные фенотипические варианты галактоземии.
Topper и соавт. в 1962 году описали вариант галактоземии у негров, a Beutler и соавт. в 1965 году представили описание варианта галактоземии Дюарте. Gitzelmann описал новое наследственное заболевание, также характеризующееся нарушением обмена галактозы — недостаточность галактокиназы, которое иногда рассматривается как один из вариантов галактоземии (так называемый швейцарский вариант). В 1972 году Gitzelmann обнаружил новый генетический блок в обмене галактозы — дефицит УДФ-галактозо-4-эпимеразы.
Разработка физико-химических методов выделения и очистки галактозо-1-фосфат-уридилтрапсферазы (Tedesco, Tedesco и соавт.) позволила провести серию исследований, направленных па выяснение иммунохимических свойств фермента у больных галактоземией.
Л. И. Ермакова и соавт. разработали систему раннего выявления и этапной диагностики галактоземии у детей, которая может быть применена в практическом здравоохранении.
Значительным достижением в области лечения аномалий обмена галактозы была разработка методов диетической коррекции метаболического дефекта. Bickel в 1954 году впервые указал на возможность использования лечебной диеты при наследственных аномалиях обмена веществ, и этот принцип в последующем был использован при лечении больных галактоземией (Л. И. Ермакова, С. М. Барашнева, Schwarz и соавт., Koch и соавт.).
- Рекомендуем далее ознакомиться со статьей "Варианты галактоземии и их причины"
Оглавление темы "Болезни обмена у детей":- Поражение печени при гомоцистинурии
- Примеры гомоцистинурии у детей
- Обмен углеводов у детей и его регуляция
- Методы обследования обмена углеводов у детей
- История открытия галактоземии
- Варианты галактоземии и их причины
- Клиника галактоземии и степени ее тяжести
- Пример галактоземии у ребенка
- Обследование при галактоземии - диагностика
- Диспансерное наблюдение за детьми с галактоземией