
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Галактоземия у новорожденных - причины, диагностика, лечение
Классическая галактоземия относится к наследственным болезням обмена веществ, связанным с нарушениями метаболизма углеводов; встречается с частотой 1 случай на 30 000-60 000 населения (в РФ — 1 случай на 36 800). Отмечаются поражение ЦНС, умственная отсталость и поведенческие проблемы вследствие гипоксии и тяжелого метаболического ацидоза.
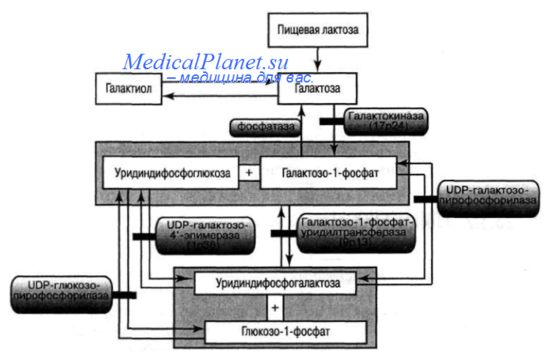
Галактоземия (классическая) — серьезное наследственное заболевание, связанное с дефектом метаболизма галактозы, обусловленным врожденным дефицитом галактозил-1-фосфат-уридилтрансферазы (или уридил-дифосфата), что приводит к накоплению в тканях токсичного для организма галактозо-1-фосфата; характеризуется тяжелым поражением печени, почек, ЦНС, органа зрения, галактозурией и альбуминурией.
F. Feillet и соавт. описали признаки катаплероза у пациента с классической неонатальной галактоземией, что подтверждает роль этого биохимического феномена в патофизиологии поражения печени у новорожденных с указанной формой галактоземии. Катаплероз — это процесс, при котором происходит удаление излишних веществ (анионов) из цикла лимонной кислоты, экспорт митохондриальных промежуточных продуктов в цитозоли, а также индукция сигнальных молекул дериватов жирных кислот.
Катаплероз относится (наряду к анаплерозом — восполнением каталитических продуктов интермедиарного обмена в цикле переноса окисляемой ацетил-КоА) к К+АТф-независимым молекулярным механизмам медиации острой регуляции высвобождения инсулина глюкозой. Катаплероз приводит к компенсации анаплероза в цикле Кребса.
Разновидности галактоземии
Помимо классической, существуют и другие формы галактоземии. В метаболизме галактозы задействованы 3 основных фермента: галактозо-1-фосфат-уридилтрансфераза (Г-1-ФУТ), галактокиназа, уридин-дифосфат-галактозо-4-эпимераза (УДФ-ГЭ).
При галактокиназной недостаточности основными метаболитами, накапливаемыми в организме, являются галактоза и галактитол. При дефиците УДФ-ГЭ патологическое накопление метаболитов происходит, как и при недостаточности Г-1-ФУТ, но при этом также отмечается нарастание уровня УДФ-галактозы в клетках и тканях. Существуют две формы УДФ-ГЭ недостаточности: доброкачественная, при которой дефицит фермента ограничивается лейкоцитами и эритроцитами (терапии не требуется), а также тяжелая (генерализованная), когда клинические проявления болезни сходны с таковыми классической (Г-1-ФУТ-дефицитной) галактоземии.
Предлагается различать три основных типа галактоземии (обозначаются римскими цифрами): недостаточность галактозо-1-фосфат-уридилтрансферазы (галактоземия I), дефицит галактокиназы (галактоземия II), недостаточность уридин-дифосфат-галактозо-4-эпимеразы (галактоземия III).
Генетические аспекты галактоземии
Галактоземия (классическая) наследуется аутосомно-рецес-сивным путем. Ген, ответственный за развитие заболевания, расположен на коротком плече хромосомы 9 (регион 9р13), а также на околоцентромерном участке хромосомы 2.
Идентифицировано около 150 генетических мутаций, приводящих к галактоземии. Наиболее частой из них у людей европеоидной расы является Q188R. Активность Г-1-ФУТ у гомозигот Q188R составляет 0%. Классическая галактоземия соответствует рубрике OMIM #230400 (по McKusick). У выходцев из других географических регионов обнаруживаются иные мутации (S135L и др.). Шесть основных мутаций (Q188R, K285N, S135L, N314D, L195P, Y209C) обнаружены у 87,5% пациентов с галактоземией.
A. Bosch и соавт. описали ряд новых мутаций при классической галактоземии: c.l52G>A/p.R51Q, c.404C>T/p.S135W, c.687G>T/p.K229N, c.756G>T/p.Q252H, c.l 140A>C/p.X380C, c.410dupT, c.821-2A>G, c.508-29delT, а также большую делецию, захватывающую экзоны 1-11.
Идентифицированы несколько неклассических (вариантных) форм недостаточности Г-1-ФУТ. Среди них наиболее частым является доброкачественный вариант Дуарте с частой мутацией N314D. При этой форме галактоземии активность Г-1-ФУТ составляет примерно 25% нормы.
Изучены мутации, приводящие к аутосомно-рецессивно наследуемой галактокиназной недостаточности. Ген, кодирующий галактокиназу, локализован на хромосоме 17 (регион 17q23-25).
В свою очередь, ген УДФ-галактозо-4-эпимеразы (галактоэпимеразы) локализуется на хромосоме 1 в регионе 1р36 (по другим данным — 1р32-р ter). J.S. Chhay и соавт. (2008) при анализе трех мутаций УДФ-галактозо-4'-эпимеразы (S81R, Т150М и P293L) обнаружили, что у пациентов, экспрессирующих аллели УДФ-ГЭ, выявляются биохимические и метаболические нарушения малой выраженности.
Клинические проявления и диагностика галактоземии
Первые проявления классической галактоземии появляются у новорожденных через несколько дней после начала вскармливания грудным молоком или его заменителями на основе коровьего молока. Отмечаются рвота, нарушения стула (диарея), снижение массы тела (или отсутствие прибавки) и задержка роста, выраженная и продолжительная желтуха (с преобладанием в крови фракции неконъюгированного билирубина), которым сопутствуют признаки интоксикации и гипогликемия. Постепенно у детей нарастает гепатолиенальный синдром (гепатоспленомегалия) с нарушением функции печени.
При отсутствии лечения наступает дисфункция почек (почечная недостаточность). По завершении неонатального периода для классической галактоземии характерны поражения ЦНС в виде диффузной мышечной гипотонии, прогрессирующей задержки психомоторного развития.
В течение первых 6 мес жизни у детей формируется катаракта, что происходит вследствие отложения и накопления в хрусталике галактитола, приводящего к разрыву зонулярных волокон. В неонатальном периоде у больных галактоземией отмечается повышенный риск заболеваемости сепсисом, вызываемым кишечной палочкой; в ряде случаев эта форма генерализованной инфекции предваряет установление диагноза галактоземии и может являться летальной.
При варианте галактоземии Дуарте (промежуточный уровень фермента) у гетерозиготных индивидов в некоторых случаях определяется до 75% галактозо-1-фосфатуридилтрансферазы. У гомозигот, несмотря на функциональную аномалию фермента, клинические проявления обычно отсутствуют, хотя иногда может отмечаться временная катаракта.
В отличие от поражения многих органов и систем, имеющих место при галактоземии с трансферазной недостаточностью, при варианте болезни, обусловленном дефицитом галактокиназы, практически единственным клиническим признаком болезни являются катаракты, которые могут в редких случаях осложняться симптомами церебрального псевдотумора.
При тяжелой галактоземии вследствие эпимеразной недостаточности к числу названных выше симптомов также относятся выраженная мышечная гипотония и глухота. К счастью, эта форма галактоземии встречается чрезвычайно редко (с частотой 1 случай на 1 млн новорожденных).
Диагноз классической галактоземии (уридилтрансферазной недостаточности) в периоде новорожденности следует заподозрить при наличии следующих признаков: иктеричность кожи, гепатомегалия, срыгивания/рвота, гипогликемия, судороги, летаргия, раздражительность, сложности со вскармливанием, плохая прибавка массы тела.
Предварительный диагноз галактоземии устанавливают по результатам исследования нескольких образцов мочи, собранных при употреблении ребенком женского или коровьего молока, а также стандартных смесей, содержащих лактозу (хроматографическое или специфическое энзиматическое исследование).
Поскольку у пациентов с галактоземией галактоза оказывает токсическое влияние на ЦНС, следует избегать нагрузочных тестов (с оральным или парентеральным ее введением).
При диагностике галактоземии проводят определение галактозы, метаболитов и галактозо1-фосфата в крови, а также исследование активности галактозо-1-фосфат-уридилтрансферазы, УДФ-галактоэпимеразы в лейкоцитах. При этом следует помнить, что увеличение количества метаболитов наблюдается при всех формах галактоземии, так же как и гипогликемия; точно идентифицировать форму заболевания можно с помощью ферментных и молекулярно-генетических методов исследований.
Световая и электронная микроскопия клеток печени выявляют жировую инфильтрацию, псевдоацинусы, постепенное формирование макронодулярного цирроза печени.
Поскольку галактоземия Дуарте ассоциирована с низкой активностью галактозо-1-фосфат-уридилтрансферазы и транзиторным повышением уровней содержания галактозы и галактозо-1-фосфата, интерпретация результатов проводимого скрининга требует дифференцировки классической галактоземии от указанного доброкачественного варианта. Скрининг N314D в качестве второго этапа неонатального скрининга галактоземии облегчает дифференциальную диагностику.
Вариант галактоземии вследствие дефицита галактокиназы подтверждается лабораторно верифицированным повышением в крови концентрации галактозы при нормальной активности трансферазы и в отсутствие галактокиназной активности в эритроцитах крови.
Диагноз тяжелой галактоземии вследствие дефицита/отсутствия эпимеразы следует заподозрить, если у ребенка на фоне симптомов классической галактоземии имеется лабораторное подтверждение нормального уровня активности трансферазы. Выявление носителей гена этой формы галактоземии возможно при помощи определения активности эпимеразы в эритроцитах крови. Существует также метод антенатальной диагностики данного варианта галактоземии, основанный на исследовании культурированных клеток амниотической жидкости (или ворсинок хориона). Применяется также специфическое исследование ДНК.
D.S. Barbouth и соавт. предлагают скрининг-обследование для выявления галактоземии у новорожденных, основанное на анализе окисления галактозы до СО2 в выдыхаемом воздухе.
Необходимо своевременное выявление галактоземии с использованием соответствующих скрининг-тестов (в родильных домах), при необходимости — генетическое консультирование и анте-/пренатальная диагностика (исследование амниотической жидкости).
Лечение галактоземии
Специфической лекарственной терапии галактоземии не существует, хотя некоторые фармакопрепараты (прогестерон, тестостерон) оказывают активирующее действие на уридин-дифосфогал-пирофосфорилазу.
В качестве симптоматических средств применяются препараты, активирующие ЦНС, сосудистые средства, антиоксиданты, гепатопротекторы и т.д.
Лечебная диета — основной метод лечения классической формы галактоземии.
- Читать "Гистидинемия у новорожденных - причины, диагностика, лечение"
Оглавление темы "Неврология новорожденных детей":- Синдром ригидного младенца (stiff baby syndrome) - причины, диагностика, лечение
- Синдром Шварца-Джампеля - причины, диагностика, лечение
- Врожденная (семейная) дисавтономия (синдром Райли-Дэя) - причины, диагностика, лечение
- Гликогеноз тип IIa (болезнь Помпе) - причины, диагностика, лечение
- Инфантильные периферические нейропатии - причины, диагностика, лечение
- Фенилкетонурия (ФКУ) у новорожденных - причины, диагностика, лечение
- Болезнь мочи с запахом кленового сиропа (БМЗКС, лейциноз) у новорожденных - причины, диагностика, лечение
- Галактоземия у новорожденных - причины, диагностика, лечение
- Гистидинемия у новорожденных - причины, диагностика, лечение
- Тирозинемия у новорожденных - причины, диагностика, лечение