
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Мукополисахаридозы. Признаки и причины мукополисахаридозов.
Мукополисахаридозы (МПС) - большая группа моногенных наследственных заболеваний, обусловленных нарушением многоступенчатого процесса ферментативного катализа гликозамингликанов (ГАГ) в лизосомах. ГАГ представляют собой сложные гетеросахара, состоящие из длинных полисахаридных цепей, построенных из остатков глюкуроновой кислоты и сульфатированного гексозамина. К гликозамингликанам относятся дерматансульфат, гепарансульфат, кератансульфат, а также хондроитин-4- и хондроитин-6-сульфат.
Впервые заболевание этой группы было описано Гурлером в 1917 году. К настоящему времени идентифицировано 10 генетических вариантов МПС. Пять из них обусловлены нарушением активности сульфатаз, четыре - гликозидаз и один вариант возникает при дефиците трансферазы.
На рисунке. представлена схема обменных превращений дерматансульфата, кератансульфата и гепарансульфата, указаны основные ферментативные блоки, обусловливающие возникновение некоторых видов МПС. Нарушение активности различных ферментов приводит к увеличению экскреции с мочой различных типов ГАГ. Повышение их концентрации дает основание для предварительного диагноза МПС и позволяет спланировать последовательность проведения энзимодиагностики.
В таблице представлены сводные данные о генетических и биохимических характеристиках при различных вариантах МПС.
Появление клинических симптомов после периода нормального развития свойственно большинству болезней накопления. Это связано с достижением критического уровня нерасщепленного субстрата в лизосомах. При исследовании биоптатов различных тканей обнаруживаются увеличенные в размере лизосомы, представляющие собой раздутые вакуоли.
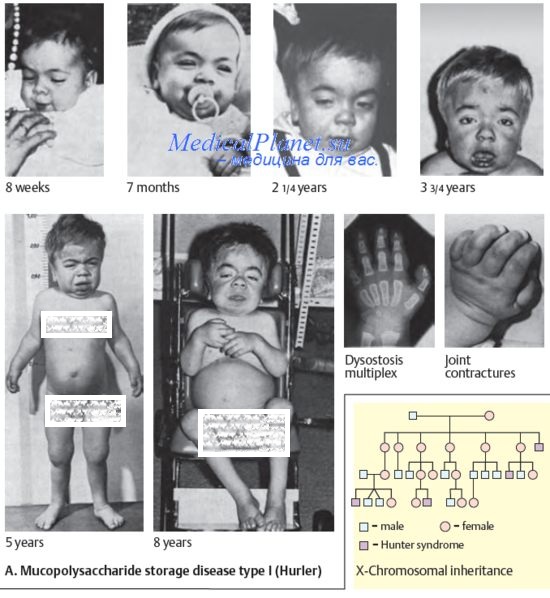
Все мукополисахаридозы наследуются аутосомно-рецессивно и лишь болезнь Хантера (МПС второго типа) наследуется Х-сцепленно рецессивно.
Клинические признаки при различных вариантах мукополисахаридозов имеют значительное сходство и становятся заметными к 2—3 летнему возрасту. Среди них: 1) грубые черты лица (гаргоилизм), 2) задержка роста (нанизм), 3) диспропорциональное строение скелета, деформации позвоночника (кифозы, сколиозы); 4) контрактуры суставов, 5) гепатоспленомегалия, 6) пахово-мошоночные грыжи, 6) снижение слуха (кондуктивная тугоухость), 7) помутнение роговицы.
Некоторые генетические варианты мукополисахаридозов сопровождаются снижением интеллекта, Сочетание отмеченных клинических признаков при разных нозологических формах этой группы заболеваний варьирует. Первый тип МПС характеризуется наличием двух аллельных вариантов -болезни Гурлера и болезни Шейе. Они обусловлены разными мутациями в одном и том же гене. Клинические проявления этих двух вариантов значительно отличаются друг от друга. Так, при МПС Гурлера у больного есть все вышеперечисленные симптомы, характерные для мукополисахаридозов в целом. Течение заболевания тяжелое, прогрессирующее; продолжительность жизни больных снижена. При синдроме Шейе симптомы, характерные для мукополисахаридозов, выражены незначительно; заболевание имеет доброкачественное течение.
Мукополисахаридоз 2-го типа (болеть Хантера) по клинической картине и тяжести течения напоминает МПС 1-го типа.
Фенотипические проявления мукополисахаридоза 3-го типа (болезни Санфилиппо) обусловлены снижением активности четырех ферментов, участвующих в процессе деградации гепарансульфата, В соответствии с этим выделяют четыре генетических формы, которые практически невозможно отличить при клиническом обследовании больного. Для этого типа МПС наиболее характерны симптомы, связанные с поражением нервной системы: снижение интеллекта, повышенная возбудимость, нарушение сна и спастическая диплегия. Как правило, у больных снижен слух. Гаргоилизм для этой формы заболевания не типичен, однако, отмечается гепатоспленомегалия и не резко выраженные изменения скелета.
Особенностью клинических проявлений мукополисахаридоза 4-го типа (болезни Моркио) являются выраженные скелетные деформации: резкое укорочение туловища, приводящее к значительному снижению роста больного, деформация позвоночника и грудной клетки, укорочение шеи, контрактуры и деформации суставов. Такие значительные деформации позвоночника нередко вызывают симптомы сдавления спинного мозга в спинно-мозговом канале. Интеллект больных, как правило, не страдает.
Клинические проявления мукополисахаридоза 6-го типа (болезни Марото-Лами) в значительной степени сходны с таковыми при МПС 1-го типа, однако, клинические симптомы появляются позднее и темп прогрессирования заболевания более медленный. Кроме того, для этого варианта МПС не характерно снижение интеллекта.
- Читать далее "I-клеточная болезнь. Сфинголипидозы. Лейкодистрофии."
Оглавление темы "Наследственные болезни обмена.":1. Альбинизм. Глазо-кожный альбинизм первого типа.
2. Галактоземия. Причина и признаки галактоземии.
3. Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов.
4. Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов.
5. Наследственные болезни обмена липидов. Причины и признаки болезни обмена липидов.
6. Несфероцитарные анемии. Недостаточность глюкозо-6-фосфат-дегидрогеназы.
7. Сфероцитарные анемии. Гемоглобинопатии.
8. Эритроцитарные мембранопатии. Анемия Минковского-Шофара. Лизосомные болезни.
9. Мукополисахаридозы. Признаки и причины мукополисахаридозов.
10. I-клеточная болезнь. Сфинголипидозы. Лейкодистрофии.