
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Галактоземия. Причина и признаки галактоземии.
В эту группу заболеваний входит несколько нозологических форм, которые обусловлены снижением активности ферментов, участвующих в метаболизме моносахаров (глюкозы, фруктозы и галактозы), дисахаров (лактозы и сахарозы) и полисахаров (гликогена). Наиболее распространенные наследственные болезни углеводного обмена - галактоземия и гликогенозы.
Это заболевание впервые описано А. Рессом в 1908 году. Его частота не превышает 1:15 000 новорожденных. Тип наследования — аутосомно-рецессивный. В настоящее время выделяют три генетических варианта, которые обусловлены недостаточностью основных ферментов, превращающих галактозу в клетках печени в глюкозу (рис. 23.5). На первом этапе метаболического пути галактоза, образованная в кишечнике из лактозы, фосфорилируется в печени под действием галактозо-1-фосфорилазы. Продукт этой реакции — галактоза-1-фосфат реагирует с уридиндифосфоглюкозой с образованием уридиндифосфогалактозы (UDP-галактоза) и глюкозо1-фосфата. Эта реакция катализируется ферментом ПФУТ. Следующий этап превращения галактозы в глюкозу происходит под действием фермента галактоэпимеразы с участием NAD+ в качестве кофермента, и приводит к образованию UDP-глюкозы. Практически все эти реакции обратимы и глюкоза может легко превращаться в галактозу, которая, таким образом, не является незаменимым пищевым компонентом.
Наиболее часто встречается галактоземия 1 -го типа, обусловленная недостаточностью ПФУТ. Ген заболевания картирован на хромосоме 9р13. Основной тип мутаций — однонуклеотидные замены. Мажорной мутацией является Gin 188Arg. В основе патогенеза заболевания лежит токсическое действие на мозг, печень, кишечник и почки галактозы и галактозо-1 -фосфата, накапливающихся в организме в результате метаболического блока. Токсическое действие заключается в ингибировании бактерицидной активности лейкоцитов, приводящем к развитию септических проявлений. При значительном накоплении галактозы она может метаболизироваться по побочному пути с образованием сахарного спирта - галактиола. Увеличение содержания этого вещества часто приводит к разрыву зонулярных волокон хрусталика и возникновению катаракт.
Клинические симптомы возникают через несколько дней после первого приема молочной пищи и характеризуются повторной рвотой, диареей и желтухой. В первые недели жизни ребенка формируется гепатомегалия и гемолитические проявления. По мере роста ребенка появляются отчетливые признаки задержки психомоторного развития, внутричерепной гипертензии, гипотрофии и почечной недостаточности. У большинства больных в первые месяцы жизни формируется катаракта. При несвоевременном лечении больные могут погибнуть от почечной или церебральной недостаточности.
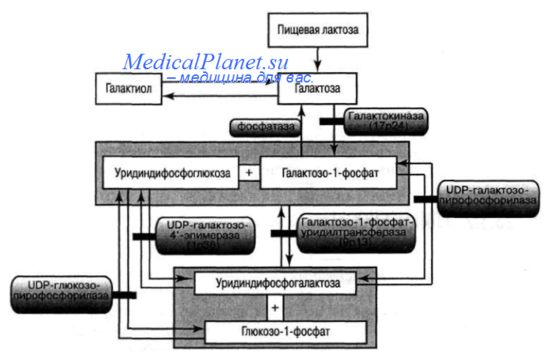
Диагностику галактоземии можно проводить на доклинической стадии путем биохимического скрининга новорожденных. В более старшем возрасте диагноз ставится на основании:
1) типичных клинических проявлений;
2) повышения концентрации галактозы и галактозо- 1-фосфата в плазме и моче и недостаточности ПФУТ в эритроцитах, лейкоцитах и фибробластах;
3) выявления мутации в гене ПФУТ.
Основной метод лечения галактоземии — безлактозная диета. Грудное молоко и молочные смеси заменяют на гидролизат казеина и соевое молоко. Необходимо отметить, что назначение безлактозной диеты не всегда приводит к полному исчезновению симптомов, как это происходит при фенилкетонурии, что связано с самоинтоксикацией больных галактозой, образующейся в организме из глюкозы.
Выявление заболевания возможно на дородовой стадии. Для этого исследуют активность фермента ПФУТ в культуре клеток плода и определяют наличие галактиола вамниотической жидкости. Возможна и прямая диагностика — выявление мутации в гене этого фермента при ДНК-анализе.
Гликогенозы - группа наследственных заболеваний, обусловленных недостаточностью ферментов, которые участвуют в синтезе и распаде гликогена. Этот полисахарид представляет собой гомополимер, имеет древовидную структуру и является основным депо глюкозы в печени и мышцах. Биосинтез гликогена происходит под действием нескольких ферментов, основные из которых: гексокиназа мышц и глюкокиназа печени, фосфоглкжомутаза и UDP-глкжозо-1 -фосфат-уридилтрансфераза. Наиболее важными ферментами деградации гликогена являются: гликоген-фосфорилаза (обладающая тканеспеиифичностью) и деветвяшие ферменты: олиго-1,4-1,4-глюкантрансфераза и амило-1,6-глкжозидаза. Под действием гликоген-фосфорилазы происходит расщепление 1 -4 гликозидньгх связей, с образованием глюкозо-1-фосфата. Олиго-1,4-1,4-глюкантрансфераза обеспечивает перенос трисахаридного фрагмента с одной цепи на другую, а амило-1,6-глюкозидаза осуществляет гидролитическое расщепление, полностью удаляющее ветвление цепей. Заключительный этап распада гликогена снова происходит под действием фосфорилазы. Совместное действие этих ферментов приводит к полному расщеплению гликогена. Снижение активности различных ферментов расщепления приводит к избыточному отложению в тканях гликогена нормальной или аномальной структуры.
К настоящему времени описано более 10 гликогенозов, но наиболее часто встречаются пять из них. В зависимости от преимущественного накопления гликогена выделяют печеночную, мышечную и генерализованную формы гликогеноза.
- Читать далее "Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов."
Оглавление темы "Наследственные болезни обмена.":1. Альбинизм. Глазо-кожный альбинизм первого типа.
2. Галактоземия. Причина и признаки галактоземии.
3. Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов.
4. Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов.
5. Наследственные болезни обмена липидов. Причины и признаки болезни обмена липидов.
6. Несфероцитарные анемии. Недостаточность глюкозо-6-фосфат-дегидрогеназы.
7. Сфероцитарные анемии. Гемоглобинопатии.
8. Эритроцитарные мембранопатии. Анемия Минковского-Шофара. Лизосомные болезни.
9. Мукополисахаридозы. Признаки и причины мукополисахаридозов.
10. I-клеточная болезнь. Сфинголипидозы. Лейкодистрофии.