
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов.
Болезнь Гирке возникает в результате снижения активности фермента глюкозо-6-фосфатазы. Существует несколько генетических вариантов, наследующихся аутосомно-рецессивно.
Первый вариант возникает в результате мутации структурного гена глюкозо-6-фосфатазы эндоплазматического ретикулума, картированного на хромосоме 17q21. Второй вариант обусловлен мутациями в гене Са-связывающего полипептида, стабилизирующего каталитическую активность глюкозо-6-фосфатазы эндоплазматического ретикулума. Локус гена не идентифицирован. Мутационное нарушение его структуры приводит к инактивации и недостаточности фермента. Третий вариант связан с мутациями структурного гена Т1-белка, обеспечивающего микросхемный транспорт глкжозо-6-фосфата. Этот ген картирован на хромосоме 1 lq23. Снижение концентрации белка Т1 также приводит к недостаточности глюкозо-6-фосфатазы. Недостаточность фермента обусловливает гипогликемию даже при малейшем голодании {из-за блокады гликогенолиза и глюконеогенеза) и накопление гликогена в печени, почках и слизистой кишечника, что приводит к дисфункции этих органов. Накопление субстрата блокированной реакции, глюкозо-6-фосфата, в свою очередь, стимулирует гликолиз и накопление лактата. Стимуляция гликолиза ведет к увеличению синтеза глицерола и ацетил-СоА, субстратов и кофакторов синтеза триглицеридов в печени.
Клинические проявления варьируют. Первые признаки заболевания появляются на первом году жизни (чаше в 3—4 месяца) и характеризуются гипогликемией, лактат-ацидозом. гепатомегалией и гипогликемическими судорогами. Больные отстают в росте и имеют низкий вес. Наблюдаются локальные отложения жира, преимущественно на щеках («кукольное» лицо), груди, ягодицах, бедрах. Moiyr возникать ксантомы на локтях, коленях, ягодицах и бедрах. Гипогликемия, чаще бессимптомная, или с судорогами и тяжелым лактат-ацидозом, возникает при малейшем голодании и при отсутствии своевременного диагноза и лечения приводит к смерти в возрасте от 1 -го года до 3-х лет. Метаболический ацидоз дает декомпенсацию и развивается респираторный дистресс-синдром при заболеваниях верхних дыхательных путей. Если больной переживает острые метаболические кризы младенческого возраста, течение заболевания становится хроническим. Прогрессирует нарушение функции почек, подагрический артрит, отставание в росте, задержка полового созревания, носовые кровотечения, остеопения, остеопороз, склонность к переломам и гепатомегалия с печеночной недостаточностью. У некоторых больных развивается легочная гипертензия и сердечная недостаточность, приводящая к смерти в юношеском возрасте.
При биохимическом исследовании выявляется лактат-ацидоз, гипогликемия. При биопсии печени отмечается недостаточность глюкозо-6-фосфатазы.
Целью лечения является поддержание нормального уровня глюкозы в крови. Показаны частые кормления в течение дня с ночными назогастральными вливаниями растворов глюкозы (8-10 мг/кг/мин у младенцев или 5-7 мг/кг/мин у детей старше 3 лет) или полный переход на парентеральное питание для купирования гипогликемии, Потребление фруктозы и галактозы должно быть ограничено, так как они не могут быть превращены в глюкозу. С возрастом гипогликемия становится менее выраженной. Как крайняя мера, при несостоятельности других методов терапии может использоваться пересадка печени и/или почек.
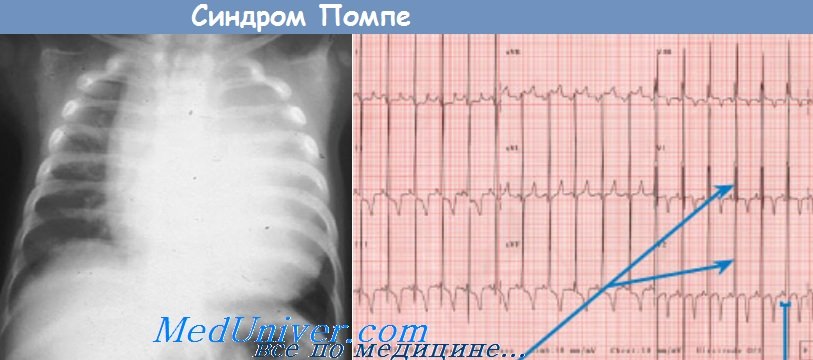
Болезнь Помпе возникает в результате мутации в гене лизосомной кислой a-D-глюкозидазы, которая обеспечивает деградацию гликогена в лизосомах. Тип наследования заболевания - аутосомно-репессивный. Ген картирован на хромосоме 17q23. Недостаточность фермента ведет к отложению негидролизованного гликогена в лизосомах мышц - сердечной и скелетных («пенистые» клетки — при морфологическом исследовании), что сопровождается картиной прогрессирующей мышечной дистрофии. Клинические проявления варьируют. Различают раннюю инфантильную, позднюю инфантильную, ювенильную и взрослые формы с хроническим течением заболевания. Ранняя инфантильная форма возникает в первые месяцы жизни и характеризуется плаксивостью, снижением двигательной активности, генерализованной прогрессирующей мышечной слабостью, включая дыхательную мускулатуру. Наблюдается задержка психомоторного развития: ребенок не держит голову, не сидит, при пальпации может выявляться гипертрофия мышц. Выявляются трудности вскармливания за счет дисфагии, гипотрофия. Характерными признаками заболевания являются макроглоссия, выраженная кардиомегалия и гепатомегалия. Смерть больных наступает в возрасте до 1 года от сердечной или сердечно-легочной недостаточности,
Поздняя инфантильная и ювенильная формы начинаются в младенчестве, в раннем и старшем детском возрасте -от 3 до 10 лет. Клинические проявления характеризуются прогрессирующей мышечной дистрофией и висцеромегалией (кардиомегалия, гепатомегалия, спленомегалия). Смерть наступает на втором десятилетии жизни от декомпенсированной сердечно-легочной недостаточности.
Взрослая форма манифестирует на 2-3 десятилетии жизни и проявляется симптомами дистальной миопатии, сколиозом грудного отдела, лордозом, медленно прогрессирующей сердечной недостаточностью. Больные доживают до старости.
- Читать далее "Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов."
Оглавление темы "Наследственные болезни обмена.":1. Альбинизм. Глазо-кожный альбинизм первого типа.
2. Галактоземия. Причина и признаки галактоземии.
3. Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов.
4. Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов.
5. Наследственные болезни обмена липидов. Причины и признаки болезни обмена липидов.
6. Несфероцитарные анемии. Недостаточность глюкозо-6-фосфат-дегидрогеназы.
7. Сфероцитарные анемии. Гемоглобинопатии.
8. Эритроцитарные мембранопатии. Анемия Минковского-Шофара. Лизосомные болезни.
9. Мукополисахаридозы. Признаки и причины мукополисахаридозов.
10. I-клеточная болезнь. Сфинголипидозы. Лейкодистрофии.