
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов.
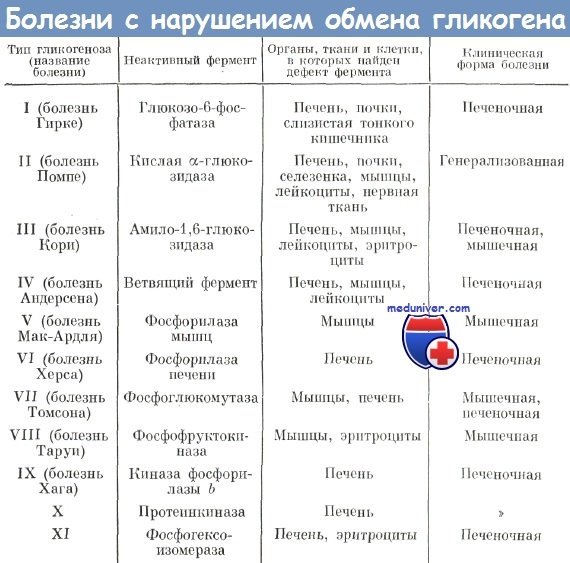
Гликогеноз третьего типа связан с мутациями структурного гена цитозольной амило-1,6-глюкозидазы, экспрессирующейся во многих тканях: печени, мышцах, эритроцитах. Ген картирован на хромосоме 1р21. Как и предыдущие варианты, третий тип гликогеноза наследуется по аутосомно-рецессивному типу. В популяции евреев-сефардов (выходцев из Северной Африки) болезнь встречается с частотой 1:5400 новорожденных. Амило-1,6-глкжозидаза участвует в метаболизме гликогена в точках ветвления гликогенового «дерева». Фермент является бифункциональным: с одной стороны, превращает лимит-декстрин в гликоген с наружными цепями нормальной длины и, с другой стороны, освобождает глюкозу путем гидролиза а-1,6-глюкозидной связи. Недостаточность фермента приводит к нарушению гликогенолиза и накоплению в тканях молекул гликогена аномальной формы с укороченными наружными цепями. Также как и при гликогенозах 1 и 2 типов, при этом варианте заболевания нарушение гликогенолиза сопровождается гипогликемией, лактат-ацидозом, гиперкетонемией.
Клинические проявления варьируют. Различают две клинические формы с хроническим течением: IIIа- при которой возникают симптомы поражения печении мышц и IIIb - при которой поражается только печень. Заболевание начинается в возрасте от 6 мес до 3-х лет. Клиническая картина в детском возрасте сходна с таковой при гликогенозе I типа. Характерные симптомы - гепатомегалия, отставание в росте, гипотрофия, «кукольное» лицо, локальные отложения жира, кожные ксантомы. Типичны лактат-ацидоз с гиперкетонемией при голодании. Гепатомегалия с дисфункцией печени, найденная у всех больных в детстве, имеет тенденцию исчезать в постпубертатный период. Больные обычно доживают до взрослого возраста, но возможен синдром внезапной смерти младенца. У взрослых больных доминирует миопатия с прогрессирующей мышечной слабостью при физической нагрузке (иногда в виде шаткой походки), гипотрофия мышц дистальных отделов нижних конечностей (преимущественно икроножных мышц), в более позднем возрасте присоединяется поражение мышц рук.
Диагностика заболевания проводится на основе клинической картины и данных лабораторного исследования: снижения активности амило- 1,6-глюкозидазы и отложения гликогена измененной структуры в гепатоцитах и мышцах. В плазме крови отмечается увеличение концентрации лактата, мочевой кислоты, холестерина и триглицеридов.
Болезнь Андерсена
Болезнь Андерсена возникает в результате мутаций гена микросомной амило-1,4:1,6-глюкантрансферазы, приводящих к ее недостаточности в печени, мышцах, лейкоцитах, эритроцитах и фибробластах. Ген картирован на хромосоме 3р 12. Тип наследования - аутосомно-рецессивный.
Амило-1,4:1,6-глюкантрансфераза участвует в синтезе гликогена в точках ветвления гликогенового дерева. Фермент соединяет сешент из, по-крайней мере, шести а-1,4-сцепленных глюкозидных остатков наружных цепей гликогена с гликогеновым «деревом» а-1,6-гликозидной связью. При недостаточности фермента в клетках печени и мышц откладывается амилопектин, что приводит к повреждению клеток. Концентрация гликогена в печени не превышает 5%.
Заболевание манифестирует на первом году жизни неспецифическими гастроин-тестинальными симптомами: рвотой, диареей. По мере ирогрессирования заболевания возникает гепатоспленомегалия, прогрессирующая печеночная недостаточность, генерализованная мышечная гипотония и атрофия, а также тяжелая кардиомиопатия. Смерть больных обычно наступает до 3-5 лет вследствие хронической печеночной недостаточности, редко — в старшем детском возрасте (до 8 лет).
Лабораторная диагностика основана на обнаружении гликогена с измененной структурой в биоптате печени и снижении активности амило-1,4:1,6-глюкантрансферазы.
Болезнь Мак-Ардля
Болезнь Мак-Ардля представляет собой мышечный вариант пшкогеноза, при котором все патологические признаки отмечаются только в мышечной ткани. В основе патогенеза лежит снижение активности мышечной гликоген-фосфорилазы, которая составляет около 5% всех растворимых мышечных белков. Ген этого фермента локализован в области хромосомы I lql3. Основной тип мутапий - однонуклеотидные замены. Мажорной считают нонсенс-мутацию в 49 кодоне первого экзона. Тип наследования — аутосомно-рецессивный.
Первые признаки заболевания возникают в детском или юношеском возрасте и характеризуются появлением болезненных мышечных спазмов после физических нагрузок, которые исчезают в период расслабления. Мышечные спазмы сопровождаются выраженным уплотнением мышц. Достаточно часто, во время тонических спазмов, могут наблюдаться выраженные вегетативные симптомы: усиление потоотделения, тахикардия, побледнение кожных покровов. По мере прогрессирования заболевания могут формироваться контрактуры крупных суставов.
Диагностика заболевания включает клиническое обследование, определение активности мышечной гликоген-фосфорилазы в биоптате мышечного волокна. Для подтверждения диагноза проводят молекулярно-генетический анализ с целью обнаружения мутаций в гене этого фермента.
- Читать далее "Наследственные болезни обмена липидов. Причины и признаки болезни обмена липидов."
Оглавление темы "Наследственные болезни обмена.":1. Альбинизм. Глазо-кожный альбинизм первого типа.
2. Галактоземия. Причина и признаки галактоземии.
3. Болезнь Гирке. Болезнь Помпе. Гликогенозы первого и второго типов.
4. Болезнь Андерсена. Болезнь Мак-Ардля. Гликогенозы третьего, четвертого, пятого типов.
5. Наследственные болезни обмена липидов. Причины и признаки болезни обмена липидов.
6. Несфероцитарные анемии. Недостаточность глюкозо-6-фосфат-дегидрогеназы.
7. Сфероцитарные анемии. Гемоглобинопатии.
8. Эритроцитарные мембранопатии. Анемия Минковского-Шофара. Лизосомные болезни.
9. Мукополисахаридозы. Признаки и причины мукополисахаридозов.
10. I-клеточная болезнь. Сфинголипидозы. Лейкодистрофии.