
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Патогенез моногенных заболеваний. Механизмы развития моногенных болезней.
Патогенез моногенных заболеваний многообразен, что связано с различиями функций белковых продуктов, нарушенных в результате мутаций. Пострадать могут функции структурных, транспортных, рецепторных и других вирусных белков. Кроме того известно, что одни белки являются тканеспецифичными и экспрессируются в определенных клетках организма (например, глобиновые белки - только в эритроцитах, а другие (например, коллагеновые белки) широко представлены в различных органах и тканях. Естественно, что клинические проявления мутаций в тканеспецифичных генах ограничены определенными тканями, в то время как при мутациях в генах широко экспрессирующихся белков наблюдается мультисистемность поражения.
Разнообразие клинических проявлений наследственного заболевания может быть связано со специфическими функциями участвующего в патогенезе белкового продукта. Белки могут играть роль ферментов, гормонов, структурных компонентов клеточного матрикса или ионных каналов, транскрипционных факторов, трасмембранных переносчиков, клеточных рецепторов, компонентов сигнальной трансдукции, иммуноглобулинов, а также выполнять другие функции, Это приводит к тому, что патогенез возникновения клинических признаков при отдельных нозологических формах в значительной степени уникален, однако, существуют и некоторые общие закономерности развития патологии. В настоящее время появилась тенденцияк выделению отдельных групп заболеваний на основании сходства патогенетических механизмов. Так, в качестве отдельной группы рассматривают каналопатии, возникновение которых обусловлено нарушением функционирования ионных каналов (калиевых, натриевых, кальциевых и хлорных) в мембране мышечных, нервных и глиальных клеток; гемоглобинопатии, возникающие при нарушении функций молекулы гемоглобина; болезни, связанные с нарушением сигнальной трансдукции, клеточных рецепторов, структуры коллагена (коллагенопатии) и др.
Наиболее хорошо изучен патогенез наследственных болезней обмена, при которых известны структура, функции и пути метаболизма белковых продуктов гена. Большинство наследственных болезней обмена — это ферментопатии, при которых нарушаются процессы расщепления и утилизации определенных продуктов.
К сожалению, классификация наследственных болезней на основании сходства патогенетических механизмов не всегда оказывается целесообразной и удобной для врача, так как в одну группу могут попасть заболевания, имеющие различные клинические проявления и, в соответствии с клинической систематикой, рассматриваемые в различных классах. Так, например, к группе болезней ионных каналов относят различные варианты наследственных эпилепсии, миотонии и пароксизмальные миоплегии, имеющие совершенно различные клинические проявления. С другой стороны, выделение заболеваний на основе общности патогенеза, облегчает процесс разработки патогенетической терапии, направленной на коррекцию общих механизмов развития болезней, В рамках данного руководства, безусловно, невозможно проанализировать все известные на сегодняшний день молекулярные механизмы наследственных патологий, но часть из них будет рассмотрена в разделах, посвященных конкретным моногенным болезням.
Аутосомно-доминантный тип наследования характеризуется: 1) проявлением заболевания у гетеро- и гомозигот по патологической мутации; 2) передачей мутантного гена от родителей к детям обоего пола с вероятностью 50%. Следует также учитывать некоторые особенности аутосомно-доминантного типа наследования заболевания в некоторых семьях: 1) неполную пенетрантность и варьирующую экспрессивность мугантного аллеля при его сегрегации в ряду поколений; 2) существование антиципации, то есть, нарастания тяжести заболевания у больных последующих поколений.
Механизм доминантности, обусловливающий появление клинических симптомов при наличии лишь одной копии мугантного аллеля, до конца не расшифрован. Эффект доминирования определенного аллеля гена может возникать в том случае, если его продукт регулирует комплекс метаболических путей, служит ключевым ферментом в ряду биохимических реакций или играет роль мембранного рецептора. При ряде заболеваний доминантный эффект может быть обусловлен нарушением синтеза белка, который участвует в формировании комплекса структур (например, белков коллагена I типа).
Варьирующая экспрессивность гена проявляется различной тяжестью клинических симптомов болезни у пораженных членов родословной. Так, у части больных могут наблюдаться стертые и абортивные формы заболевания, для обнаружения которых необходимо использовать дополнительные методы обследования, в то время как удругих - наблюдаются тяжелые клинические проявления.
Неполная пенетрантность гена выявляется при анализе родословных при наличии так называемого «пропуска поколений», когда передача заболевания может осуществляться через клинически здорового носителя патологического гена, который он унаследовал от больного родителя. При наличии неполной пенетрантности мугантного гена риск возникновения заболевания у потомков пораженного родителя может быть модифицирован и рассчитан по формуле: р x Р, где р — априорная вероятность возникновения заболевания, предполагаемая на основе менделевских закономерностей (50% при аутосомио-доминантном типе наследования), а Р - коэффициент пенетрантности. Показатель пенетрантности может быть суммарным, полученным при анализе большого числа информативных родословных или рассчитанным на основании изучения родословной единственной семьи. В последнем случае он рассчитывается на основании соотношения заболевших носителей мугантного гена к общему количеству носителей (заболевших и не заболевших) среди членов одной семьи. Примеры родословных при заболеваниях с полной и неполной пенетрантностью представлены на рисунке.
Механизмы возникновения антиципации недостаточно ясны, однако один из них был расшифрован при изучении болезней, обусловленных экспансией тринуклеотидных повторов.
Необходимо иметь в виду, что только часть случаев заболеваний с аутосомно-доминантным типом наследования обусловлена передачей патологического гена родителями, имеющими этот ген в каждой клетке организма. Определенная доля аутосомно-доминантных заболеваний обязана своим появлением вновь возникшей мутации в единственной половой клетке одного из родителей. Для каждого заболевания количество случаев, обусловленных новой мутацией и сегрегацией мугантного гена в ряду поколений варьирует. Их соотношение связано, прежде всего, с различиями в приспособленности больного индивида. Для заболеваний, начинающихся в старшем возрасте, не приводящих к выраженной инвалидизации, не сопровождающихся значительными пороками развития, снижением продолжительности жизни и фертильности больных, наибольшее количество случаев, как правило, являются сегрегирующими. Если приспособленность больных в силу ряда причин снижена (раннее начало, быстрая инвалидизация, тяжелые врожденные пороки развития, снижение продолжительности жизни больного), преобладающее число случаев заболевания обусловлено вновь возникшей мутацией (de novo). Так, при ахондроплазии, характерный признак которой - непропорциональная карликовость, 80% случаев возникает в результате мутации de novo в гаметах родителей. Мужчины, страдающие этим заболеванием как правило бесплодны, фертильность пораженных женщин тоже снижена. С другой стороны основное количество случаев хореи Гентингтона — тяжелого неиродегенеративного заболевания, сопровождающегося выраженными гиперкинезами, деменцией и психическими нарушениями — являются сегрегирующими, что связано с поздним возрастом начала болезни и нормальной фертильно-стью носителей гена на доклинической стадии. Важно отметить, что эта закономерность прослеживается не всегда. Так при ряде заболеваний с относительно доброкачественным течением может наблюдаться преобладание случаев, обусловленных новой мутацией, что объясняется повышенной частотой мутирования того или иного гена. К таким заболеваниям относится, например, наследственная моторно-сенсорная нейропатия 1А-типа, для которой характерна умеренная выраженность клинических симптомов, наличие большого количества стертых форм болезни и неполная пенетрантность. Заболевание не приводит к выраженной инвалидизации и снижению плодовитости больных, однако, не менее 70% случаев этого заболевания обусловлено мутацией de novo. Еще одна интересная особенность этой болезни заключается в довольно редком типе мутации — дупликации хромосомной области, содержащей дозочувствительный ген. В настоящее время известно мало заболеваний, возникновение которых обусловлено дупликациями того или иного гена. Показано, что повышение уровня белкового продукта таких генов приводит к возникновению клинических симптомов в результате нарушения гомеостаза. На примере наследственной моторно-сенсорной нейропатии 1А-типа продемонстрируем один из возможных механизмов возникновения эффекта доминантности при наличие мутации в гетерозиготном состоянии.
- Читать далее "Болезнь Шарко-Мари-Тута. Наследственная мотосенсорная нейропатия."
Оглавление темы "Болезни с менделирующим типом наследования.":1. Генетическая гетерогенность. Клиника моногенных болезней.
2. Патогенез моногенных заболеваний. Механизмы развития моногенных болезней.
3. Болезнь Шарко-Мари-Тута. Наследственная мотосенсорная нейропатия.
4. Болезнь ионных каналов. Характеристика болезни ионных каналов.
5. Коллагенопатии. Признаки и диагностика коллагенопатий.
6. Аутосомно-рецессивный тип наследования. Характеристика аутосомно-рецессивных заболеваний.
7. Муковисцидоз. Кистозный фиброз. Диагностика муковисцидоза.
8. Проксимальная спинальная амиотрофия. Болезнь Вердниг—Гофмана.
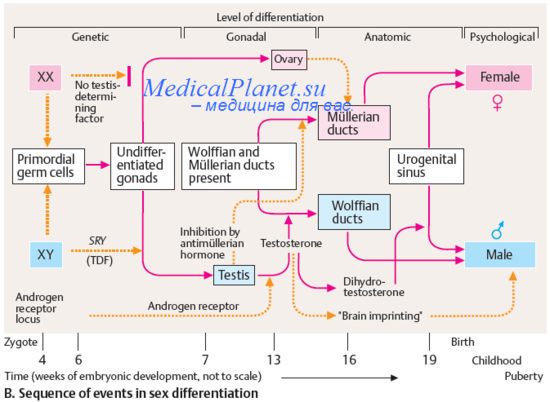
9. Нарушения половой дифференцировки. Адреногенитальный синдром.
10. Мужские нарушения половой дифференцировки. Синдром персистенции мюллеровых протоков.