
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины семейной гиперхолестеринемии и механизмы ее развития
Семейная гиперхолестеринемия является болезнью, развивающейся вследствие мутации в гене, кодирующем рецептор к ЛПНП, который участвует в транспорте и метаболизме холестерина. В результате нарушается обратная связь и повышается уровень холестерина, что приводит к развитию раннего атеросклероза, а следовательно, повышает риск развития инфаркта миокарда.
Семейная гиперхолестеринемия — одно из наиболее распространенных заболеваний, наследуемых по законам Менделя. Частота гетерозигот с одним мутантным геном в популяции составляет 1 случай на 500 человек. С рождения у таких людей уровень холестерина повышен в 2-3 раза, что приводит к формированию сухожильных ксантом и раннему атеросклерозу во взрослом возрасте.
Гомозиготы с двумя копиями мутантного гена имеют значительно более тяжелые повреждения, поскольку у них уровень холестерина повышен в 5-6 раз. У этих пациентов развитие кожных ксантом и атеросклеротическое поражение коронарных, церебральных и периферических сосудов происходит уже в молодом возрасте, а инфаркт миокарда может развиться до 20 лет. Крупномасштабные исследования выявили, что семейную гиперхолесте-ринемию имеют 3-6% лиц, перенесших инфаркт миокарда.
Для понимания этой патологии необходимо вспомнить процессы нормального метаболизма и транспорта холестерина. Приблизительно 7% холестерина тела циркулируют в плазме в основном в форме ЛПНП. Количество холестерина ЛПНП плазмы зависит от скорости его синтеза и катаболизма; ключевую роль в этих процессах играет печень. На первом этапе синтеза ЛПНП из печени в кровь поступают липопротеины очень низкой плотности (ЛПОНП). ЛПОНП богаты триглицеридами, однако они содержат меньше эфиров холестерина.
В капиллярах жировой ткани и мышц частицы ЛПОНП подвергаются липолизу ферментом липопротеинлипазой, в результате большинство триглицеридов высвобождаются. Образовавшиеся молекулы липопротеинов промежуточной плотности (ЛППП) содержат небольшое количество триглицеридов, но богаты эфирами холестерина и сохраняют на своей поверхности два из трех ЛПОНП-ассоциированных апопротеинов — В-100 и Е. После высвобождения из эндотелия капилляров метаболизм ЛППП может идти двумя путями. Около 50% синтезированных ЛППП поступают обратно в печень с помощью рецепторов к ЛПНП.
Рецепторы, связывающие ЛППП на мембране гепатоцитов, распознают и апопротеин В-100, и апопротеин Е. Эти рецепторы называют рецепторами к ЛПНП, т.к. они участвуют в расщеплении ЛПНП до ЛПОНП в печени. Оставшиеся вне клеток печени ЛППП подвергаются дальнейшему метаболизму: триглицериды и апопротеин Е удаляются, в результате формируются богатые холестерином ЛПНП. Необходимо отметить, что ЛППП — первый и основной источник ЛПНП.
Считается, что существует два механизма удаления ЛПНП из плазмы: с помощью рецептора к ЛПНП и с помощью рецептора к окисленным ЛПНП (скавенджер-рецептора, который будет описан далее; от англ. scavenger — уборщик.). Несмотря на то что рецепторы к ЛПНП обнаруживаются на поверхности многих типов клеток, в т.ч. на фибробластах, лимфоцитах, гепатоцитах, клетках гладких мышц и адренокортикальных клетках, около 70% ЛПНП плазмы метаболизируются печенью посредством сложной транспортной системы. Сначала ЛПНП связываются с поверхностными рецепторами, которые сгруппированы в специализированных участках на плазматической мембране, называемых окаймленными ямками (кавеолами). После этого участки с рецепторами, связанные с ЛПНП, интернализируются путем инвагинации с образованием окаймленных пузырьков, которые мигрируют внутрь клетки и сливаются с лизосомами.
На этом этапе ЛПНП открепляются от рецепторов, которые снова переносятся на мембрану. В лизосомах молекулы ЛПНП подвергаются ферментативному расщеплению, при этом апопротеин гидролизуется до аминокислот, а эфиры холестерина разрушаются с образованием свободного холестерина. Свободный холестерин сквозь мембрану лизосом поступает в цитоплазму клетки, где используется для синтеза липидов мембран и регуляции гомеостаза холестерина. Для высвобождения холестерина из лизосом необходимы два белка — NPC1 и NPC2 (см. «Болезнь Ниманна-Пика типа С»).
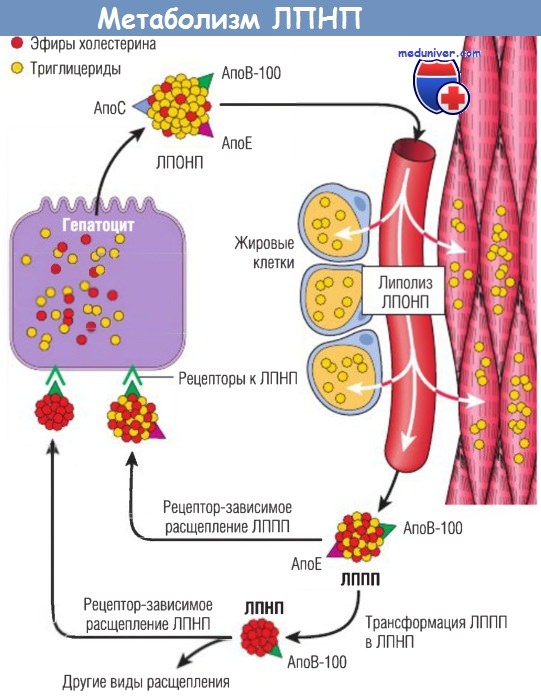
Липолиз липопротеинов очень низкой плотности (ЛПОНП) липопротеинлипазой приводит к высвобождению в капилляры триглицеридов,
которые впоследствии откладываются в адипоцитах и используются скелетными мышцами как источник энергии.
АпоВ-100 — апопротеин В-100; АпоС — апопротеин С; АпоЕ — апопротеин Е; ЛППП —липопротеины промежуточной плотности.
Внутриклеточный холестерин влияет на следующие процессы, не связанные между собой:
- подавляет синтез холестерина за счет ингибирования активности фермента 3-гидрокси-3-метилглутарил-коэнзим А-редуктазы, который является фактором, ограничивающим скорость синтеза холестерина;
- активирует фермент ацил-коэнзим А: холестерин-ацилтрансферазу, способствующий этерификации и хранению избыточного холестерина;
- подавляет синтез поверхностных рецепторов к ЛПНП, защищая клетку от избыточного накопления холестерина.
Как упоминалось ранее, семейная гиперхолестеринемия развивается вследствие мутации в гене рецепторов к ЛПНП. Для гетерозигот характерно наличие 50% от нормального количества высокоаффинных рецепторов к ЛПНП, т.к. гетерозиготы имеют только один нормальный аллель этого гена. Результатом такой недостаточности транспорта является нарушение рецептор-зависимого пути катаболизма ЛПНП, вследствие чего приблизительно в 2 раза увеличивается уровень ЛПНП плазмы крови. У гомозигот нормальный рецептор на поверхности клеток практически не встречается, а уровень циркулирующих ЛПНП значительно выше нормы.
Кроме того, для гомо- и гетерозигот характерно увеличение синтеза ЛПНП, механизм которого также связан с дефицитом рецепторов к ЛПНП. Увеличение синтеза ЛПНП вносит свой вклад в патогенез семейной гиперхолестеринемии. Вспомните, что для транспорта в печень ЛППП, непосредственного предшественника ЛПНП плазмы крови, на поверхности гепатоцитов также необходимы рецепторы к ЛПНП (к апопротеинам В-100 и Е). При семейной гиперхолестеринемии нарушение транспорта ЛППП в печень вторично увеличивает пул ЛППП плазмы крови, являющихся предшественниками ЛПНП плазмы.
Считается, что транспорт ЛПНП с помощью ска-венджер-рецепторов по крайней мере в некоторой степени осуществляется в клетки системы монону-клеарных фагоцитов. Моноциты и макрофаги имеют рецепторы к химически модифицированным (например, ацетилированным или окисленным) ЛПНП. В норме количество ЛПНП, транспортируемых с помощью скавенджер-рецепторов, меньше, чем количество ЛПНП, переносимых с помощью рецепторов к ЛПНП. Однако при семейной гиперхолестеринемии наблюдается значительное увеличение транспорта холестерина в мононуклеарные фагоциты и в стенки сосудов с помощью скавенджер-рецепторов. Это приводит к появлению ксантом на коже и преждевременному атеросклерозу.
Молекулярная генетика семейной гиперхолестеринемии чрезвычайно сложна. Было выявлено более 900 мутаций в гене рецептора к ЛПНП, в т.ч. вставки, делеции, миссенс- и нонсенс-мутации. Эти мутации могут быть разделены на пять категорий. Мутации I класса встречаются относительно редко, они приводят к полной утрате синтеза рецептора (нуль-аллель). При II классе мутаций, наиболее распространенной форме, рецепторный белок накапливается в эндоплазматическом ретикулуме вследствие нарушенного фолдинга и невозможности транспортировки в аппарат Гольджи.
Мутации III класса поражают связывающий ЛПНП домен рецептора. Такие рецепторы транспортируются на поверхность клетки, однако не способны к нормальному связыванию с ЛПНП. Мутации IV класса приводят к образованию рецепторов, которые транспортируются на поверхность клеток и нормально связываются с ЛПНП, однако группируются не в кавеолах и, следовательно, не интернализируются после соединения с ЛПНП. Мутации V класса кодируют рецепторы, способные к транспорту на поверхность клетки, взаимодействию с ЛПНП и интернализации, однако pH-зависимого расщепления рецептора и связанного с ним ЛПНП не происходит. Такие рецепторы остаются в эндосомах и разрушаются, вместо того чтобы вернуться на поверхность клетки.
Выявление ключевой роли рецепторов к ЛПНП в регуляции гомеостаза холестерина привело к разработке группы препаратов, которые снижают уровень холестерина плазмы за счет увеличения количества рецепторов к ЛПНП. Одна из терапевтических стратегий основана на способности некоторых препаратов (статинов) подавлять синтез внутриклеточного холестерина за счет снижения активности 3-гидрокси-3-метилглутарил-коэнзим А-редуктазы, усиливая таким образом синтез рецептора к ЛПНП.
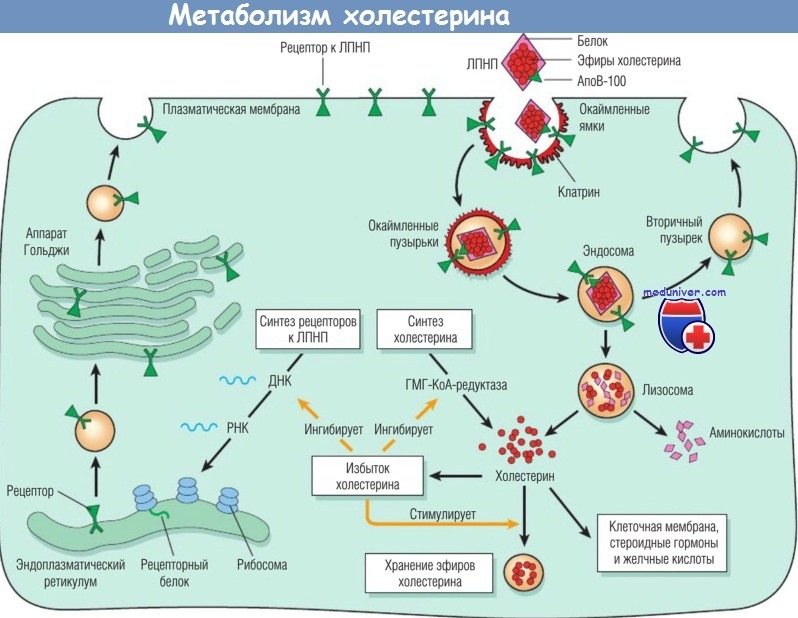
АпоВ-100 — апопротеин В-100; ГМГ-КоА-редуктаза — 3-гидрокси-3-метилглутарил-коэнзим А-редуктаза;
ДНК — дезоксирибонуклеиновая кислота; РНК — рибонуклеиновая кислота.
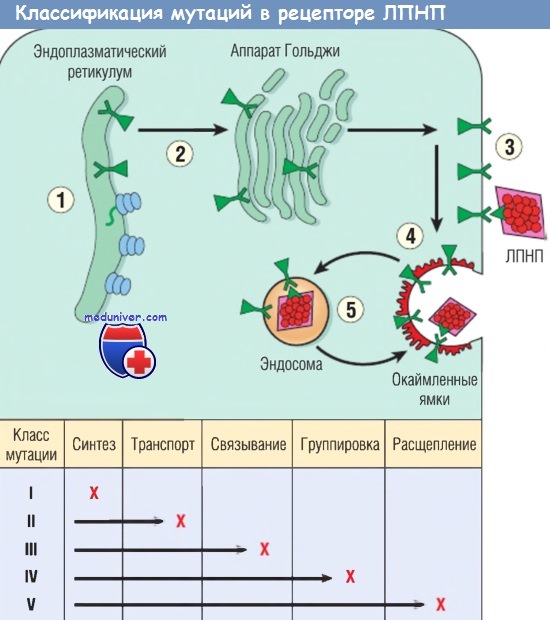
Эти мутации нарушают синтез рецептора в эндоплазматическом ретикулуме (1),
транспорт рецептора в аппарат Гольджи (2), связывание с апопротеиновыми лигандами (3), группировку в окаймленных ямках (4) и расщепление в эндосомах (5).
Для каждого класса характерна гетерогенность на уровне ДНК.
- Рекомендуем ознакомиться со следующей статьей "Причины лизосомных болезней накопления и их варианты"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития