
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины гликогенозов (болезней накопления гликогена) и механизмы их развития
Гликогенозы (или болезнь накопления гликогена) развиваются из-за наследственного дефекта одного из ферментов, участвующих в синтезе или расщеплении гликогена. В зависимости от локализации специфического фермента в норме отложение гликогена при этих заболеваниях может обнаруживаться в отдельных тканях, в большинстве органов или быть системным.
Значимость специфического дефицита фермента удобнее рассматривать с точки зрения нормального метаболизма гликогена. Гликоген является депо глюкозы. Синтез гликогена начинается с превращения глюкозы последовательно в глюкозо-6-фосфат и глюкозо-1-фосфат с помощью ферментов глюкокиназы (гексокиназы) и фосфоглюкомутазы соответственно.
Глюкозо-1-фосфат, в свою очередь, превращается в уридиндифосфоглюкозу. Далее строится чрезвычайно крупный (с молекулярной массой около 100 млн) ветвящийся полимер, содержащий до 10 тыс. молекул глюкозы, связанных друг с другом а-1,4-гликозидными связями. Основные цепи и ответвления гликогена продолжают удлиняться за счет добавления молекул глюкозы при участии гликогенсинтетазы. При деградации отдельные фосфорилазы печени и мышц отщепляют глюкозо-1-фосфат от гликогена до тех пор, пока на каждом ответвлении не останется около четырех остатков глюкозы. Образующийся в результате этого процесса разветвленный олигосахарид называют остаточным декстрином.
Этот продукт далее может расщепляться только с помощью фермента, удаляющего боковые цепи. В дополнение к основным путям гликоген также может разрушаться кислой мальтазой в лизосомах. Если в лизосомах этого фермента нет, содержащийся в них гликоген недоступен для деградации цитоплазматическими ферментами, такими как фосфорилаза.
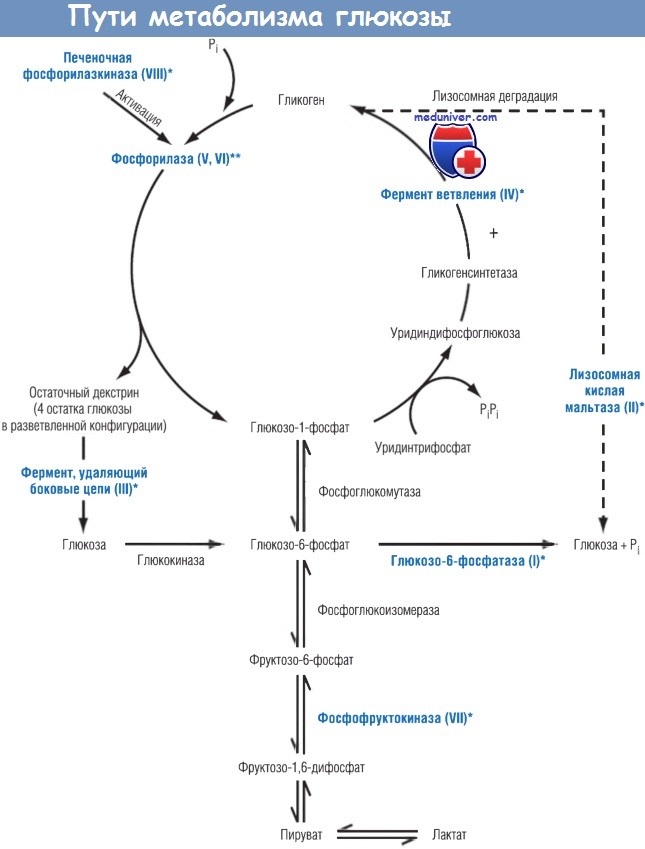
Звездочками обозначены ферментативные нарушения, приводящие к развитию болезней накопления гликогена.
Римские цифры обозначают тип болезни накопления гликогена, связанный с дефицитом конкретного фермента.
Типы V и VI развиваются вследствие дефицита мышечной и печеночной фосфорилаз соответственно. Р1—фосфатидилинозитол.
На основании специфического дефицита фермента и соответствующей клинической картины были выделены 12-13 различных типов гликогенозов, обозначенных римскими цифрами. Список, однако, продолжает расти. На основании патофизиологических механизмов эти типы заболеваний можно объединить в три группы:
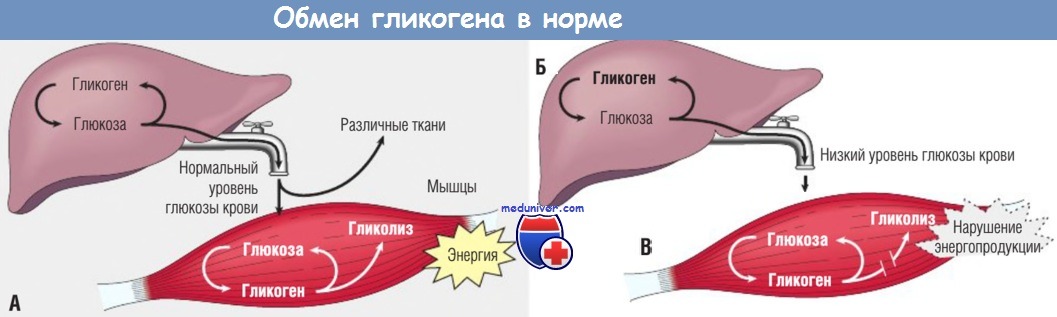
- печеночная группа. Печень играет ключевую роль в метаболизме глюкозы, поскольку содержит несколько ферментов, которые синтезируют гликоген для его отложения, а также разрушают его до свободной глюкозы, которая выделяется в кровь. Таким образом, наследственный дефицит печеночных ферментов, вовлеченных в процесс деградации гликогена, приводит не только к накоплению его в печени, но и к снижению концентрации глюкозы в крови (гипогликемии).
Наиболее типичным примером печеночной формы гликогеноза является болезнь Гирке (гликогеноз типа I), которая возникает из-за дефицита глюкозо-6-фосфатазы. Другими примерами являются дефицит печеночной фосфорилазы и фермента, удаляющего боковые цепи, которые вовлечены в процесс деградации гликогена. При каждом из этих заболеваний гликоген откладывается во многих органах, однако в клинической картине доминируют гепатомегалия и гипогликемия;
- мышечная группа. В поперечнополосатых мышцах гликоген является важным источником энергии во время физической активности. Во время гликолиза генерируется АТФ и образуется лактат. При дефиците ферментов, которые участвуют в гликолизе, в мышцах накапливается гликоген и нарушается энергопродукция, что приводит к мышечной слабости. К этой группе относят дефициты мышечной фосфорилазы (болезнь МакАрдла, гликогеноз типа V), мышечной фосфофруктокиназы (гликогеноз типа VII) и некоторые другие. Типичными симптомами мышечных гликогенозов являются судороги мышц и отсутствие повышения уровня лактата в крови после физической нагрузки, что связано с остановкой гликолиза;
- смешанная группа. Это болезни накопления гликогена, связанные с недостаточностью а-глюкозидазы (кислой мальтазы) и фермента, отвечающего за ветвление гликогена, ассоциируются с отложением гликогена во многих органах и ранней смертью. Кислая мальтаза представляет собой лизосомный фермент, недостаточность которого приводит к накоплению гликогена в лизосомах во всех органах (болезнь Помпе, гликогеноз типа II). Наиболее важным симптомом является кардиомегалия.

- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития