
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины болезни Тея-Сакса и механизмы ее развития
Ганглиозидозы GM2 представляют собой группу из трех заболеваний, вызываемых невозможностью катаболизировать ганглиозиды GM2. Для деградации ганглиозидов GM2 необходимы три полипептида, которые кодируются различными генами. Фенотипические проявления мутаций в этих генах очень схожи, т.к. приводят к накоплению ганглиозидов GM2.
Дефекты ферментов, однако, отличаются. Болезнь Тея-Сакса — наиболее распространенная форма ганглиозидозов, которая развивается вследствие мутации в гене а-субъединицы на 15-й хромосоме. Результатом такой мутации становится тяжелый дефицит фермента гексозаминидазы А. Эта болезнь наиболее распространена среди евреев, особенно восточноевропейского происхождения (ашкенази), среди которых частота носителей составляет 1 случай на 30 человек.
а) Морфология. Поскольку гексозаминидаза А отсутствует практически во всех тканях, ганглиозиды GM2 накапливаются во многих органах (например, в сердце, печени, селезенке), однако основные клинические признаки связаны с поражением нейронов центральной и вегетативной нервной системы, а также сетчатки. При гистологическом исследовании выявляются нейроны, увеличенные в размерах за счет цитоплазматических вакуолей, каждая из которых представляет собой растянутую лизосому, заполненную ганглиозидами.
Эти ткани дают положительную реакцию при окрашивании на жиры (масляным красным О и суданом черным). При электронной микроскопии можно обнаружить несколько видов цитоплазматических включений, наиболее распространенные — лизосомы, имеющие извитую форму и множество слоев мембран, что делает их похожими на луковичную кожицу. Со временем разрушение нейронов прогрессирует, наблюдаются избыточная пролиферация микроглии и накопление комплексных липидов в фагоцитах вещества головного мозга.
Аналогичный процесс происходит в мозжечке и нейронах базальных ганглиев, ствола головного мозга, спинного мозга, а также в ганглиях задних корешков и нейронах вегетативной нервной системы. Ганглионарные клетки сетчатки (особенно в районе краев макулы) также увеличены в размерах за счет накопления в них ганглиозидов GM2. Вишнево-красное пятно в макуле, окруженное желтовато-серым ободком, контрастирует с бледными раздутыми ганглионарными клетками всей остальной сетчатки. Этот симптом характерен для болезни Тея-Сакса и других болезней накопления, поражающих нейроны.
б) Клинические признаки. Больные дети при рождении выглядят нормальными, первые признаки и симптомы появляются в возрасте около 6 мес. Для заболевания характерно ухудшение умственного и психомоторного развития, начинающееся с дискоординации движений, отставания в умственном развитии с последующим появлением мышечной слабости, слепоты и прогрессированием деменции.
Практически у всех пациентов (иногда на ранних стадиях болезни) появляется вишнево-красное пятно в макуле сетчатки — характерный, но не патогномоничный симптом заболевания. За 1-2 года достигается полное вегетативное состояние; смерть наступает в возрасте 2-3 лет. В гене а-субъединицы обнаружено более 100 мутаций, большинство из которых нарушают фолдинг белка. Такие неправильно свернутые белки вызывают «ответ развернутого белка», который приводит к апоптозу.
Эти данные позволили разработать метод терапии шаперонами для лечения болезни Тея-Сакса.
Антенатальная диагностика возможна с помощью ферментативного и основанного на ДНК анализа. Две другие формы ганглиозидозов GM2 — болезнь Сандхоффа (развивающаяся вследствие дефекта b-субъединицы) и вариант АВ (недостаточность белка-активатора) — имеют сходные клинические признаки.
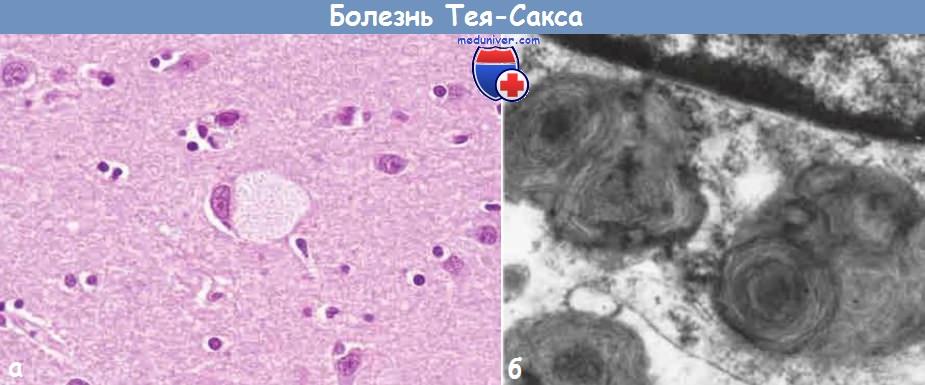
(А) Под световым микроскопом видно, что крупный нейрон содержит липидные вакуоли.
(Б) При электронной микроскопии в части нейрона видны лизосомы извитой формы. Сверху — часть ядра.
- Рекомендуем ознакомиться со следующей статьей "Причины болезни Ниманна-Пика и механизмы ее развития"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития