
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
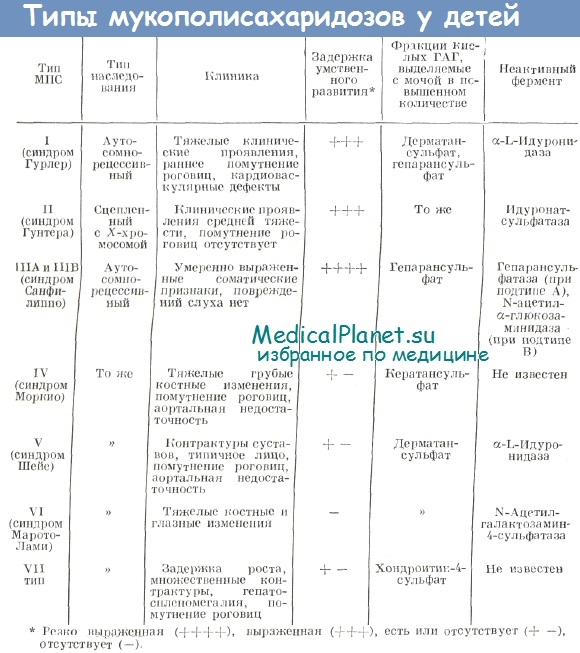
Причины мукополисахаридозов и механизмы их развития
Мукополисахаридозы представляют собой группу близких синдромов, которые развиваются вследствие генетически обусловленных дефектов лизосомных ферментов, участвующих в деградации мукополиса-харидов (глюкозаминогликанов).
Химически мукополисахариды представляют собой длинноцепочечные углеводные комплексы, которые связываются с белками с образованием протеогликанов. Их особенно много в основном веществе соединительной ткани. Среди накапливающихся мукополисахаридов можно выделить дерматансульфат, гепарансульфат, кератансульфат и хондроитинсульфат.
Ферменты, вовлеченные в процесс деградации этих молекул, отщепляют концевые сахара от полисахаридных цепей, располагающихся вдоль полипептидной цепи или корового белка. В отсутствие соответствующих ферментов эти цепи накапливаются в лизосомах различных тканей и органов.
Было описано несколько клинических групп мукополисахаридозов (I—VII), причиной которых стал дефицит определенного фермента. Все мукополисахаридозы, кроме одного, наследуются по аутосомно-рецессивному типу; исключением является синдром Гунтера, который передается потомству по сцепленному с Х-хромосомой типу.
В каждой группе (например, в группе мукополисахаридозов I, характеризующихся дефицитом a-L-идуронидазы) существуют подгруппы, которые выделили на основании различных мутантных аллелей в одном и том же генетическом локусе. Таким образом, тяжесть ферментной недостаточности и клиническая картина часто могут различаться даже среди подгрупп.
Мукополисахаридозы представляют собой прогрессирующие заболевания, признаками и симптомами которых могут быть огрубление черт лица, помутнение роговицы, ограничение подвижности суставов и умственная отсталость. Часто наблюдается увеличение экскреции с мочой накапливающихся мукополисахаридов.
а) Морфология. Мукополисахариды чаще всего системно накапливаются в мононуклеарных фагоцитах, эндотелии, клетках интимы гладких мышц и фибробластах. Наиболее часто поражаются селезенка, печень, костный мозг, лимфатические узлы, кровеносные сосуды и сердце.
При микроскопическом исследовании видно, что пораженные клетки растянуты и имеют светлую цитоплазму — клетки-баллоны. При электронной микроскопии светлая цитоплазма представлена множеством мелких вакуолей — раздутых лизосом, содержащих крошечные гранулы, окрашивающиеся PAS и биохимически являющиеся мукополисахаридами. Аналогичные лизосомные изменения обнаруживаются в нейронах при таких синдромах, для которых характерно поражение ЦНС.
В дополнение к этому некоторые лизосомы в нейронах замещаются пластинчатыми зеброподобными тельцами, сходными с теми, которые встречаются при болезни Ниманна-Пика. Общими признаками всех мукополисахаридозов являются гепатоспленомегалия, деформации скелета, патологии клапанов сердца, субэндотелиальные отложения в артериях и поражение головного мозга.
При длительном течении заболевания субэндотелиальные очаги приводят к ишемии миокарда, поэтому инфаркт миокарда и сердечная недостаточность являются частыми причинами смерти таких больных.
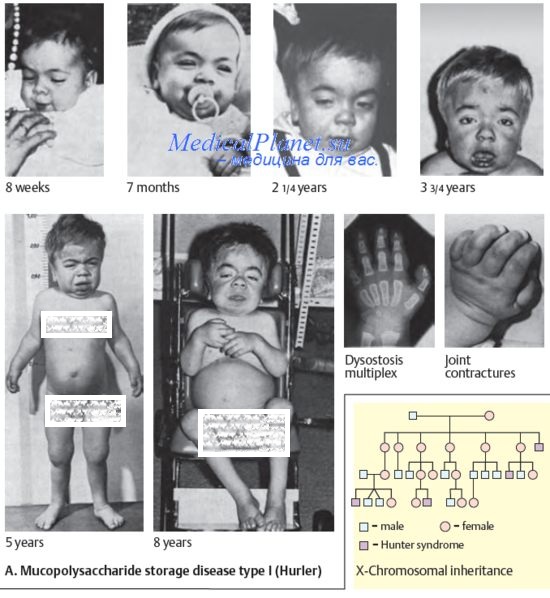
б) Клинические признаки. Из 7 в этой главе будут описаны только 2 наиболее изученные группы мукополисахаридоза. Синдром Гурлера, также известный как мукополисахаридоз I-H, развивается вследствие дефицита a-L-идуронидазы. Этот синдром — один из наиболее тяжелых мукополисахаридозов.
При рождении дети кажутся здоровыми, однако уже к 6-24 мес жизни у них развивается гепатоспленомегалия, обнаруживается задержка роста и, как и при большинстве других мукополисахаридозов, происходит огрубление черт лица, а также деформация скелета. Смерть в возрасте 6-10 лет часто наступает из-за осложнений со стороны сердца. Синдром Гунтера, также известный как мукополисахаридоз II, отличается от синдрома Гурлера механизмом наследования (сцепленное с Х-хромосомой), отсутствием помутнения роговицы и более мягким клиническим течением.
- Рекомендуем ознакомиться со следующей статьей "Причины гликогенозов (болезней накопления гликогена) и механизмы их развития"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития