
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины болезни Ниманна-Пика и механизмы ее развития
Болезнь Ниманна-Пика типов А и В характеризуется первичным дефектом кислой сфингомиелиназы и, как результат, накоплением сфигномиелина. Тип А представляет собой тяжелую инфантильную форму с обширным поражением ЦНС, выраженным висцеральным накоплением сфингомиелина, прогрессирующим истощением организма и летальным исходом в течение первых трех лет жизни.
При типе В больные, напротив, страдают органомегалией, а симптомы поражения ЦНС обычно отсутствуют. Эти пациенты, как правило, доживают до взрослого возраста. Как и болезнь Тея-Сакса, болезнь Ниманна-Пика типов А и В часто встречается среди евреев ашкенази.
Ген кислой сфингомиелиназы располагается на хромосоме 11p15.4 и является одним из генов, подвергающихся имприн-тингу. Этот ген преимущественно экспрессируется с материнской хромосомы в результате эпигенетического подавления отцовского гена. В гене кислой сфингомиелиназы было обнаружено более 100 мутаций. Считается, что есть связь между типом мутации, тяжестью ферментной недостаточности и фенотипом.
а) Морфология. При классическом инфантильном типе А миссенс-мутация приводит к полному отсутствию сфингомиелиназы. Сфингомиелин является обязательным компонентом клеточных мембран (в т.ч. мембран органелл). Дефект ферментов блокирует деградацию липидов, приводя к их прогрессирующему накоплению внутри лизосом, особенно в клетках системы мононуклеарных фагоцитов.
Вследствие накопления сфингомиелина и холестерина пораженные клетки увеличиваются в размере, достигая иногда 90 мкм в диаметре. Множество мелких вакуолей приблизительно одинакового размера придает цитоплазме пенистый вид. Замороженные срезы свежей ткани дают положительную реакцию при окрашивании на жиры.
При электронной микроскопии выявляется, что вакуоли представляют собой переполненные вторичные лизосомы, которые часто содержат мембранозные цитоплазматические тела, внешне напоминающие концентрические слоистые фигуры миелина, или, как их часто называют, зеброподобные тельца.
Наполненные липидами пенистые фагоциты встречаются в большом количестве в селезенке, печени, лимфатических узлах, костном мозге, миндалинах, ЖКТ и легких. Поражение селезенки обычно приводит к ее значительному увеличению (иногда в 10 раз по сравнению с нормой). Гепатомегалия обычно не так значительна. Лимфатические узлы по всему организму увеличены до средних или больших размеров.
Поражение головного мозга и глаз заслуживает особого внимания. Извилины головного мозга уменьшены в размерах, а борозды расширены. Присутствует диффузное поражение нейронов всех отделов нервной системы. Основной гистологической находкой является вакуолизация и вздутие нейронов, что со временем приводит к смерти клетки и утрате вещества мозга. У 30-50% пациентов обнаруживается вишнево-красное пятно на сетчатке, аналогичное пятну при болезни Тея-Сакса.
б) Клинические признаки. Клинические проявления болезни Ниманна-Пика типа А могут присутствовать уже при рождении и практически всегда становятся очевидными к 6-му месяцу жизни. Характерный признак — выпирание живота в связи с гепатомегалией. Сразу после появления первых симптомов наблюдается прогрессирование болезни: ребенок плохо набирает вес, типичны рвота, лихорадка и генерализованная лимфаденопатия в сочетании с прогрессирующим нарушением психомоторных функций. Смерть обычно наступает в течение первого или второго года жизни.
Диагноз ставят на основании биохимического анализа активности сфингомиелиназы в биоптате печени или костного мозга. Выявление носительства (и болезни) можно проводить с помощью анализа ДНК.
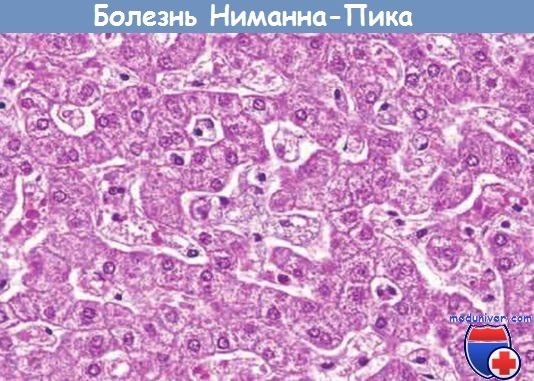
Гепатоциты и клетки Купфера придают цитоплазме пенистый, вакуолизированный вид вследствие отложения липидов.
Болезнь Ниманна-Пика типа С
Ранее болезнь Ниманна-Пика типа С считали сходной с типами А и В, но сейчас известно, что на биохимическом и молекулярном уровнях тип С значительно отличается от типов А и В. Кроме того, тип С встречается гораздо чаще, чем типы А и В, вместе взятые.
Причиной болезни Ниманна-Пика типа С являются мутации в двух генах — NPC1 и NPC2, причем поражение гена NPC1 встречается в 95% случаев. В отличие от других лизосомных болезней накопления, болезнь Ниманна-Пика типа С возникает в результате первичного нарушения транспорта липидов.
Пораженные клетки накапливают холестерин и ганглиозиды Gm1 и Gm2. Кодируемые NPC1 и NPC2 белки вовлечены в процесс транспорта свободного холестерина из лизосом в цитоплазму. Болезнь Ниманна-Пика типа С клинически гетерогенна. Она может проявляться водянкой плода, мертворождением, неонатальным гепатитом, а также существовать в хронической форме, характеризующейся прогрессирующим поражением нейронов.
Болезнь Ниманна-Пика типа С впервые проявляется в детстве и характеризуется атаксией, нарушением движения глаз в вертикальном направлении, дистонией, дизартрией и психомоторной регрессией.
- Рекомендуем ознакомиться со следующей статьей "Причины болезни Гоше и механизмы ее развития"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития