
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Биохимические и молекулярные основы болезней
Как известно, моногенные (менделевские) заболевания являются результатом поражения одного гена.
Генетический дефект может приводить к образованию аномального белка или уменьшению количества белкового продукта гена. При моногенных заболеваниях различные механизмы поражения могут затрагивать практически любые типы белков. Как уже отмечалось, механизм наследования заболевания в некоторой степени связан с типом пораженного мутацией белка.
Для удобства механизмы, лежащие в основе развития моногенных заболеваний, можно разделить на четыре категории:
(1) дефекты ферментов и их последствия;
(2) дефекты мембранных рецепторов и транспортных систем;
(3) нарушения структуры, функции или количества неферментных белков;
(4) генетически детерминированные побочные реакции на лекарственные средства.
а) Дефекты ферментов и их последствия. Мутации могут приводить к синтезу дефектного фермента со сниженной активностью или к уменьшению количества нормального фермента. В обоих случаях последствием является метаболический блок. На рисунке ниже приведен пример ферментативной реакции, в которой субстрат превращается в конечный продукт через промежуточные продукты 1 и 2 с помощью внутриклеточных ферментов.
В этой реакции конечный продукт регулирует фермент 1 посредством обратной связи. Существует также дополнительный путь, синтезирующий небольшие количества метаболита 1 (M1) и метаболита 2 (М2). Биохимические эффекты дефекта ферментов в такой реакции могут приводить к следующим основным последствиям:
- накопление субстрата в зависимости от этапа, на котором происходит блок, может сопровождаться накоплением одного или обоих промежуточных продуктов. Накопление промежуточного продукта 2 может стимулировать дополнительный путь и таким образом приводить к избытку M1 и М2. При этом, если предшественник, промежуточные или конечный продукты дополнительного пути являются токсичными в высоких концентрациях, происходит повреждение тканей.
Например, при галактоземии дефицит галактозо-1-фосфатуридилтрансферазы приводит к накоплению галактозы и, как результат, поражению тканей. Избыточное накопление комплексных субстратов в лизосомах вследствие недостаточности разрушающих ферментов обусловливает развитие заболеваний, обычно называемых лизосомными болезнями накопления,
- дефект фермента может приводить к метаболическому блоку или снижению количества конечного продукта, необходимого для нормального функционирования клетки. Например, дефицит меланина может быть результатом недостатка тирозиназы, которая нужна для биосинтеза меланина из его предшественника тирозина. Этот дефицит ассоциируется с клиническим состоянием, известным как альбинизм.
Если конечный продукт воздействует на ферменты, участвующие в начальных реакциях, посредством отрицательной обратной связи, следствием дефицита конечного продукта может стать избыток промежуточных продуктов и продуктов их катаболизма, некоторые из которых могут быть токсичными в высоких концентрациях. Примером заболевания с таким механизмом развития является синдром Леша-Нихана;
- неспособность инактивировать повреждающий ткани субстрат наилучшим образом иллюстрирует дефицит альфа-1-антитрипсина. Организм лиц, страдающих наследственной формой дефицита альфа-1-антитрипсина в сыворотке, не способен инактивировать нейтрофильную эластазу в легких. Бесконтрольная активность этой протеазы приводит к разрушению эластина в стенках альвеол и, как следствие, к развитию эмфиземы легких.
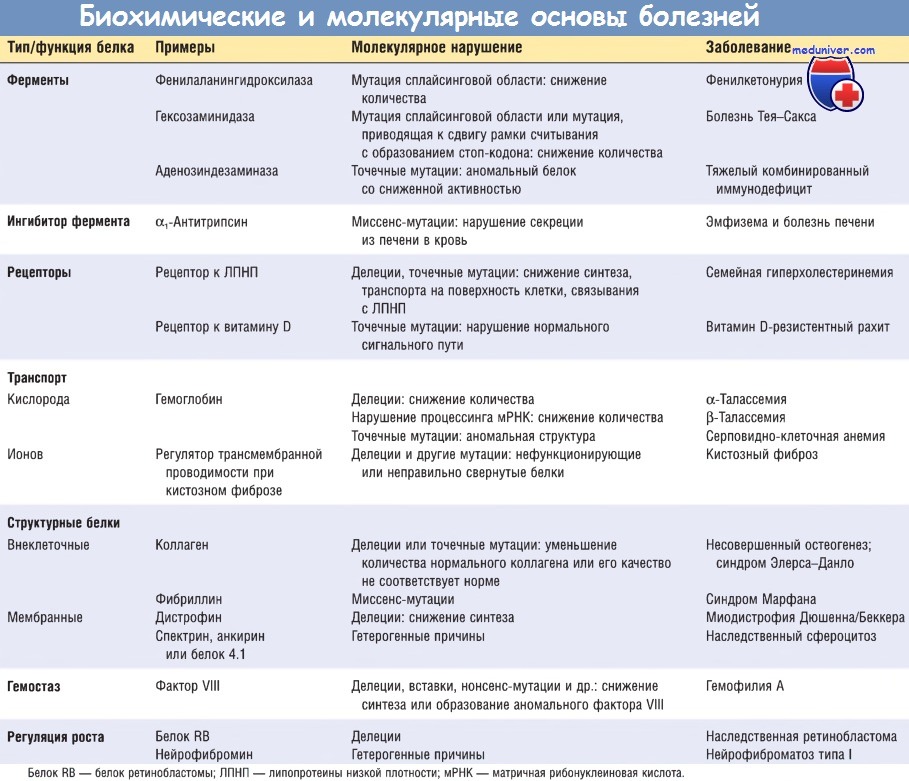
б) Дефекты мембранных рецепторов и транспортных систем. Многие биологически активные субстраты активно транспортируются через клеточную мембрану, в большинстве случаев посредством одного из двух механизмов: рецептор-зависимого эндоцитоза или транспортного белка.
Примером генетического дефекта рецептор-зависимой транспортной системы может служить семейная гиперхолестеринемия, при которой уменьшение синтеза или функции рецепторов к ЛПНП приводит к нарушению транспорта ЛПНП в клетки и вторичному чрезмерному синтезу холестерина за счет сложных промежуточных механизмов.
При кистозном фиброзе дефект обнаруживается в транспортной системе ионов хлора в экзокринных железах, потовых протоках, легких и поджелудочной железе. Нарушение транспорта ионов хлора приводит к тяжелым повреждениям легких и поджелудочной железы; механизм этих повреждений еще до конца не изучен.
в) Нарушения структуры, функции или количества неферментных белков. Генетические нарушения, приводящие к изменениям в неферментных белках, часто имеют распространенные вторичные эффекты. Пример — серповидно-клеточная анемия. Гемоглобинопатии, одной из которых является серповидно-клеточная анемия, характеризуются нарушением структуры молекулы глобина.
Эта группа заболеваний хорошо иллюстрирует данную категорию нарушений. Талассемии, в отличие от гемоглобинопатий, являются результатом мутации в гене глобина, что влияет на количество синтезируемых цепей глобина. Талассемии связаны с пониженным содержанием структурно нормальных цепей а-глобина или бета-глобина. Другими примерами генетических дефектов структуры служат молекулярные нарушения структуры белков коллагена, спектрина и дистрофина, что приводит к несовершенному остеогенезу, наследственному сфероцитозу и мышечным дистрофиям соответственно.
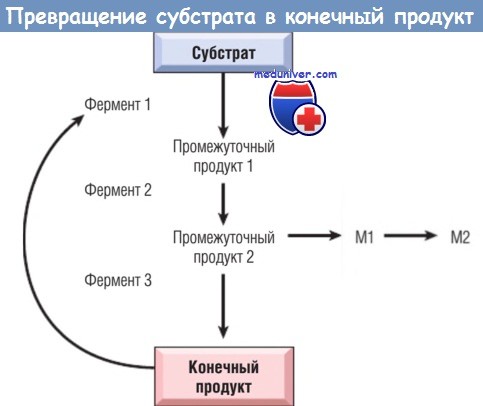
М1, М2 — метаболиты дополнительного пути.
г) Генетически детерминированные побочные реакции на лекарственные средства. Некоторые генетически детерминированные дефекты ферментов удается выявить только после того, как их носитель подвергается лечению определенными лекарственными средствами. Этот особый раздел генетики, называемый фармакогенетикой, имеет особое клиническое значение.
Классическим примером такого генетически обусловленного повреждения является состояние, связанное с дефицитом фермента глюкозо-6-фосфатдегидрогеназы. В нормальных условиях дефицит этого фермента не имеет никаких клинических проявлений, однако при введении, например, антималярийного препарата примахина развивается тяжелая форма гемолитической анемии. За последнее время было выявлено большое количество полиморфизмов генов метаболизирующих лекарственные средства ферментов, транспортеров и рецепторов.
В некоторых случаях генетические факторы имеют ключевое значение в формировании чувствительности к лекарственным средствам и побочных реакций. Предполагается, что развитие фармакогенетики позволит разработать схемы индивидуальной терапии для каждого конкретного пациента, т.е. медицина станет персонализированной.
После краткого рассмотрения биохимических основ моногенных заболеваний перейдем к описанию конкретных примеров, сгруппированных по дефектам генома.
- Рекомендуем ознакомиться со следующей статьей "Причины синдрома Марфана и механизмы его развития"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития