
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины лизосомных болезней накопления и их варианты
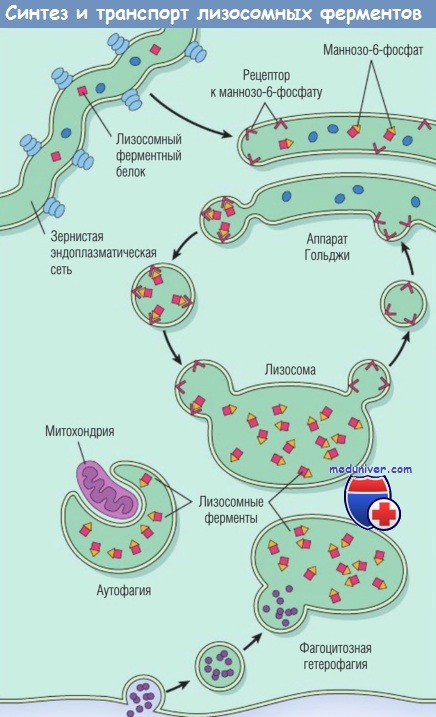
Лизосомы являются ключевым компонентом внутриклеточного пищеварительного процесса. Они содержат ряд гидролитических ферментов, которые имеют две основные функции: поддерживают кислую среду внутри лизосом и образуют особую группу белков, предназначенных для внутриклеточных органелл. Для реализации данных функций необходима специальная обработка этих белков в аппарате Гольджи.
Аналогично всем остальным секреторным белкам лизосомные ферменты (или кислые гидролазы, как их часто называют) синтезируются в эндоплазматическом ретикулуме, а затем транспортируются в аппарат Гольджи, где подвергаются различным посттрансляционным модификациям, одну из которых необходимо рассмотреть отдельно.
Эта модификация заключается в присоединении концевых маннозо-6-фосфатных групп к некоторым олигосахаридным боковым цепям. Фосфорилированные остатки маннозы служат «адресными бирками», которые распознаются специфическими рецепторами на внутренней поверхности мембраны аппарата Гольджи. Лизосомные ферменты связываются с этими рецепторами и отделяются от остальных секреторных белков аппарата Гольджи.
Затем небольшие транспортные пузырьки, содержащие связанные с рецепторами ферменты, отщепляются от аппарата Гольджи и сливаются с лизосомами. Так ферменты попадают внутрь клетки, а пузырьки возвращаются обратно к аппарату Гольджи. Как уже отмечалось, генетические нарушения этого уникального механизма сортировки белков могут привести к развитию лизосомных болезней накопления.
Кислые гидролазы катализируют разрушение различных сложных макромолекул, которые могут образовываться при переработке внутриклеточных органелл (аутофагия) или захватываться клеткой извне с помощью фагоцитоза (гетерофагия). При наследственной недостаточности лизосомного фермента катаболизм его субстрата оказывается незавершенным, что приводит к накоплению внутри лизосом частично разрушенных нерастворимых метаболитов.
Эти органеллы, наполненные не полностью переваренными макромолекулами, становятся чрезмерно большими и многочисленными и вызывают нарушения в функционировании клеток, приводя к лизосомным болезням накопления. К этим болезням также может приводить отсутствие какого-либо белка, необходимого для нормальной работы лизосом, например:
- белка, активирующего или защищающего фермент;
- белка — активатора субстрата. В некоторых случаях белки, реагирующие с субстратом для активации его гидролиза, могут быть дефектными;
- транспортных белков, необходимых для эвакуации переваренного материала из лизосом.
Существует три основных подхода к лечению лизосомных болезней накопления. Первый — заместительная терапия. Второй подход — уменьшение количества субстрата — основывается на том факте, что если уменьшить количество субстрата, то существующей активности фермента будет достаточно, чтобы разрушить и предотвратить накопление субстрата.
В основе третьего современного подхода лежит понимание молекулярных основ ферментной недостаточности. При многих заболеваниях (в частности, при болезни Гоше) ферментативная активность снижена в связи с тем, что мутантный белок нестабилен и имеет тенденцию к распаду, вследствие чего и разрушается в эндоплазматическом ретикулуме.
При таких заболеваниях экзогенный конкурентный ингибитор фермента может парадоксальным образом связываться с мутантным ферментом и служить «структурной основой» для нормального фолдинга, предотвращая таким образом его разрушение. Такую молекулярную терапию шаперонами в настоящее время тщательно исследуют.
В группу лизосомных болезней накопления включены несколько отличающихся друг от друга состояний.
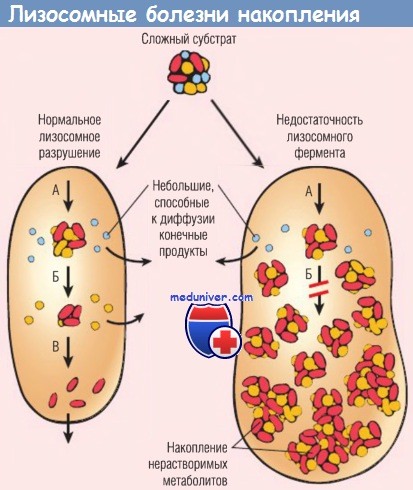
Сложный субстрат в норме разрушается набором лизосомных ферментов (А, Б и В) до растворимых конечных продуктов.
При недостаточности или неправильном функционировании одного из этих ферментов (например, Б) катаболизм является незавершенным, и в лизосомах накапливаются нерастворимые промежуточные продукты.
Распределение накапливаемого материала и, следовательно, поражение органов зависят от двух взаимосвязанных факторов:
(1) ткани, где находится больше всего материала, который необходимо разрушать;
(2) места, где это разрушение происходит в норме. Например, головной мозг богат ганглиозидами. Недостаточность гидролиза ганглиозидов, которая развивается при ганглиозидозах GM1 и GM2, в первую очередь приводит к накоплению ганглиозидов внутри нейронов, что сопровождается неврологической симптоматикой.
Нарушения деградации мукополисахаридов обусловливают поражение практически всех органов, т.к. эти вещества находятся во всех тканях организма.
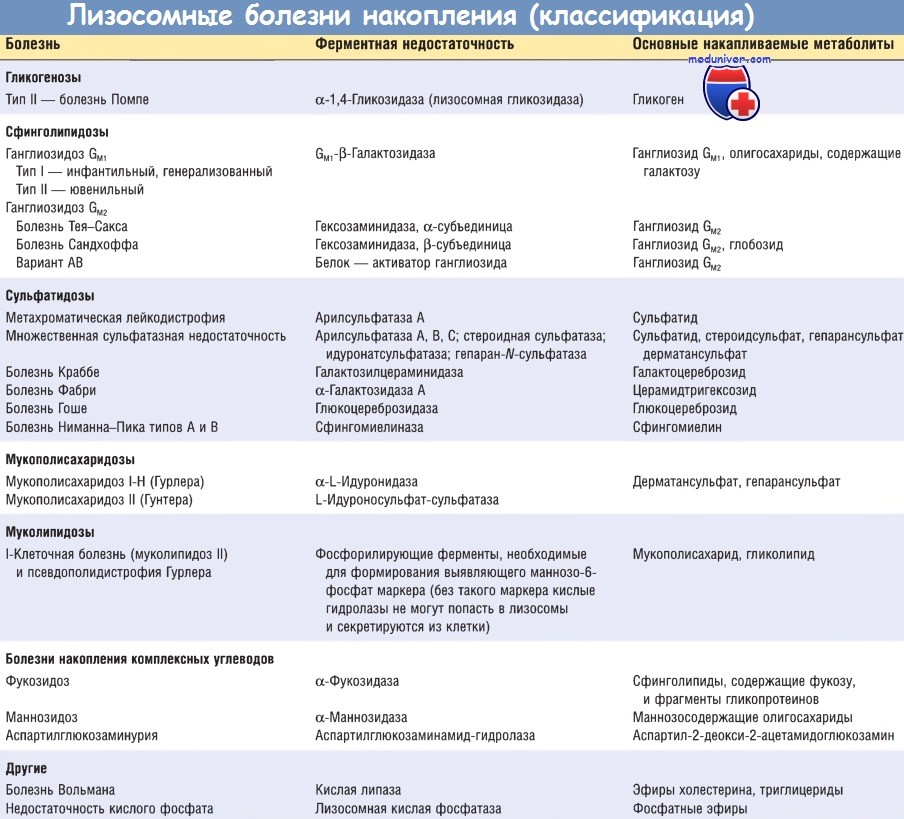
Клетки системы мононуклеарных фагоцитов особенно богаты лизосомами и участвуют в деградации множества субстратов, поэтому при лизосомных болезнях накопления происходит увеличение размеров органов, содержащих большое количество фагоцитов (например, печени и селезенки). На основании биохимической природы накапливаемых метаболитов лизосомные болезни накопления можно разделить на следующие подгруппы: гликогенозы, сфинголипидозы (липидозы), сульфатидозы, мукополисахаридозы и муколипидозы.
Далее в отдельных статьях рассмотрены наиболее распространенные заболевания из этих подгрупп.
- Рекомендуем ознакомиться со следующей статьей "Причины болезни Тея-Сакса и механизмы ее развития"
Оглавление темы "Патофизиология болезней обмена":- Биохимические и молекулярные основы болезней
- Причины синдрома Марфана и механизмы его развития
- Причины синдрома Элерса-Данло (СЭД) и механизмы его развития
- Причины семейной гиперхолестеринемии и механизмы ее развития
- Причины лизосомных болезней накопления и их варианты
- Причины болезни Тея-Сакса и механизмы ее развития
- Причины болезни Ниманна-Пика и механизмы ее развития
- Причины болезни Гоше и механизмы ее развития
- Причины мукополисахаридозов и механизмы их развития
- Причины гликогенозов (болезней накопления гликогена) и механизмы их развития