
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема нарушения обмена углеводов
Нарушение метаболизма углеводов обычно обусловлено энзимопатиями или аномалией регуляции активности ферментов.
Галактоза образуется в кишечнике при расщеплении лактозы и превращается (преимущественно в печени) в глюкозу и гликоген. Галактоземия развивается при дефекте галактозо-1-уридилтрансферазы. При поступлении лактозы с грудным молоком во многих органах накапливается галактозо-1-фосфат, который ингибирует ферменты, участвующие в метаболизме глюкозы.
Избыток галактозо-1-фосфата превращается в токсичный спирт галактитол. Галактозо-1-фосфат и галактитол оказывают токсическое действие на ЦНС, печень, почки и хрусталик глаза. Ранняя диагностика и диета, не содержащая галактозы, предотвращают повреждение органов и тканей. При этом синтез уридиндифосфат-галактозы (УДФ-галактозы) по-прежнему сохраняется. Дефицитгалоктокиназы, при котором выявляется гипергалактоземия и галактозурия, характеризуется менее серьезными нарушениями функции органов.
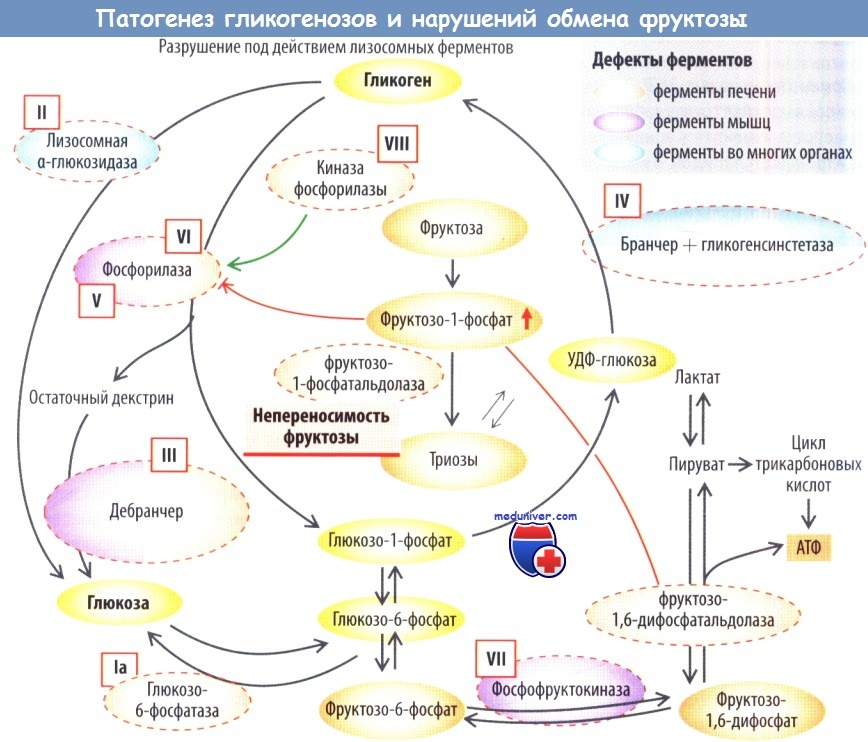
Наследственная непереносимость фруктозы обусловлена дефектом фруктозо-1-фос-фатальдолазы. Катаболизм фруктозы (моносахарид, содержащийся во фруктах) замедляется, накапливается фруктозо-1-фосфат, который в печени ингибирует гликогенфосфорилазу и фруктозо-1,6-дифосфатальдолазу.
В результате развивается гепатогенная гипогликемия, острая печеночная недостаточность или цирроз. При ранней диагностике и исключении из диеты фруктозы заболевание не сокращает продолжительность жизни, в то время как поступление фруктозы с пищей приводит к быстрой смерти вследствие печеночной недостаточности.
Болезни накопления гликогена (гликогенозы). Гликоген — депонированная форма глюкозы в организме. Синтез гликогена происходит в печени и мышцах. При распаде гликогена образуется глюкоза, которая используется в качестве источника энергии в этих тканях или поступает в другие органы. Дефекты ферментов катаболизма гликогена вызывают его накопление в органах с развитием гипогликемии.
При недостаточности ферментов распада гликогена развиваются следующие заболевания:
- тип Ia (болезнь фон Гирке, дефицит глюкозо-6-фосфатазы);
- тип Ib (дефицит микросомной глюкозо-6-фосфат-транслоказы (на схеме не показано]);
- тип II (болезнь Помпе, дефицит лизосомнойа-гликозидазы);
- тип III (болезнь Форбса, болезнь Кори, дефицит дебранчера — фермента, препятствующего ветвлению, наиболее распространенный тип);
- тип V (болезнь Мак-Ардла, дефицит мышечной фосфорилазы);
- тип VI (болезнь Герса, дефицит печеночной фосфорилазы);
- тип VIII (болезнь Томсона, дефицит печеночной фосфорилазы b-киназы).
Дефицит синтеза гликогена (очень редко) приводит к гликогенозу типа IV (болезнь Андерсена, дефицит бранчера — ветвящегося фермента). Синтезируется необычный тип гликогена, который накапливается в мозге, сердце, мышцах и печени. Тип VII (болезнь Таури, дефицит мышечной фосфофруктокиназы) характеризуется нарушением распада глюкозы и уменьшением образования энергии для мышечной ткани.
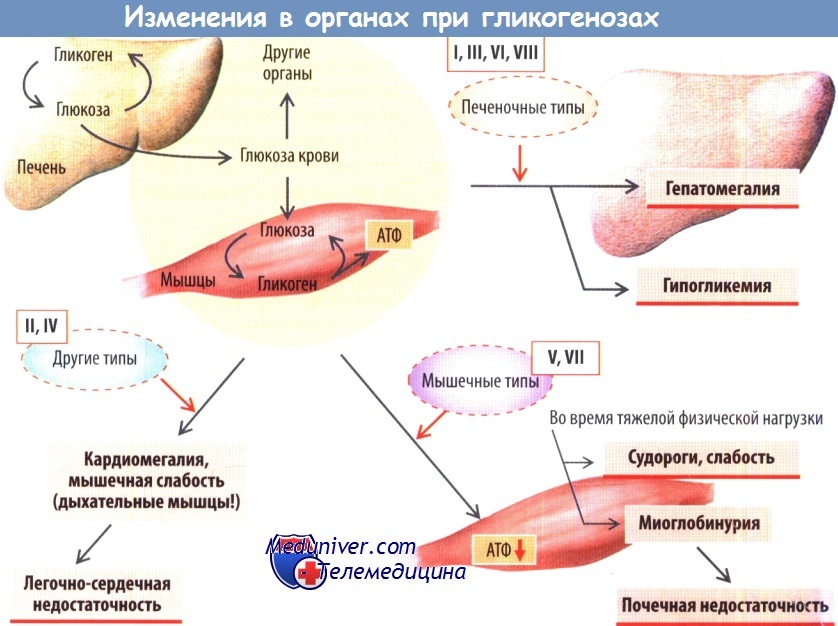
В зависимости от локализации первичных нарушений, связанных с дефицитом фермента, выделяют печеночные (I, III, VI, VIII), мышечные (V, VII) и другие типы (II, IV) гликогенозов. К основным симптомам печеночных типов гликогенозов относятся гепатомегалия вследствие избыточного накопления гликогена и гипогликемия, при мышечных гликогенозах — дефицит энергии.
Физическая нагрузка у больных с гликогенозом не сопровождается повышением уровня лактата в сыворотке, вызывает быструю утомляемость, мышечные судороги и боль, а также миоглобинурию (при V типе), которая может привести к почечной недостаточности. Кардиомегалия, слабость дыхательных мышц при гликогенозе II типа и печеночная недостаточность при гликогенозе IV типа часто приводят к смерти в детском возрасте.
- Рекомендуем ознакомиться со следующей статьей "Схема нарушения обмена жиров (липидозов)"
Оглавление темы "Схемы патогенеза нарушений метаболизма":- Схема нарушения обмена аминокислот
- Схема нарушения обмена углеводов
- Схема нарушения обмена жиров (липидозов)
- Схема нарушения обмена липопротеидов
- Схема развития подагры
- Схема развития гемохроматоза
- Схема развития болезни Вильсона
- Схема последствий дефицита α-1-антитрипсина
- Схема развития диспротеинемий (нарушения обмена белков)
- Схема развития порфирии