
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема развития гемохроматоза
Железо (Fe) — важнейший элемент, необходимый для синтеза молекул гемоглобина (Hb) в эритроцитах и миоглобина в мышцах. Железо также входит в состав цитохромов и других ферментов. Кроме того, Fe играет важную роль в вирулентности бактерий. Образование комплексов Fe с белками (лактоферрином, сидерокалином, липокалином, некоторыми белками острой фазы) относится к механизмам защиты против патогенных микроорганизмов.
Около 25 % Fe депонируются в комплексе с белками. Белок ферритин находится в клетках слизистой кишечника, печени, костном мозге, эритроцитах, плазме;имеет«карман», который связывает 4500 ионов Fe3+ на 1 молекулу. Железо ферритина высвобождается легче (приблизительно 600 мг), чем железо гемосидерина (250 мг в макрофагах печени и костного мозга).
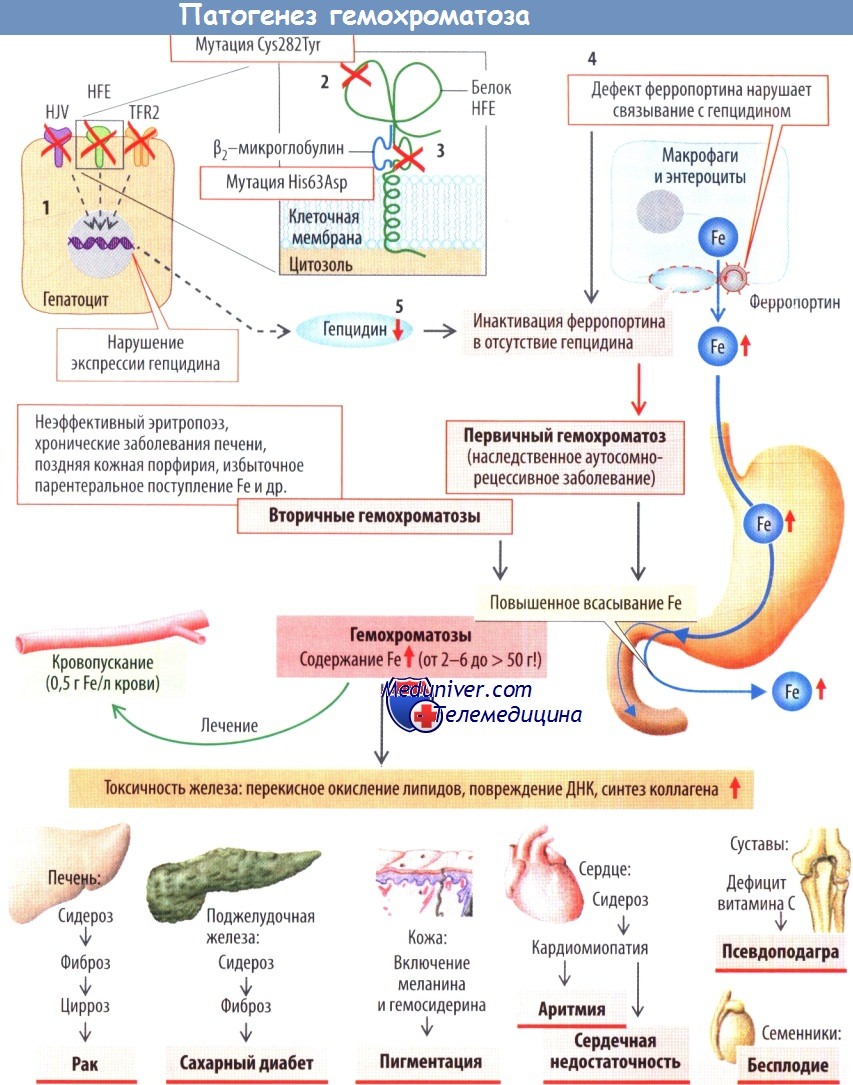
Дефицит Fe вызывает анемию, а избыток Fe может способствовать окислительному повреждению клеток. Поэтому гомеостаз железа жестко регулируется, включая процессы всасывания, рециркуляции, заполнения или опустошения депо Fe. Основную роль в регуляции этих процессов играет печеночный пептидный гормон гепцидин. Его экспрессия усиливается при избытке Fe и по механизму обратной связи ингибируется при дефиците Fe.
В регуляции экспрессии гепцидина участвуют белок HFE, рецептор трансферрина 2-го типа (TFR2), гемоювелин (HJV). Синтез гепцидина повышается при воспалении (стимулируется IL-6) и избытке Fe (стимулируется железом трансферрина); снижается при гипоксии (стимулируется эритропоэз) и дефиците Fe. Активация экспрессии гепцидина происходит под действием ма-триптазы-2 — сериновой протеазы, связанной с клеточной мембраной, которая расщепляет гемоювелин.
Гемохроматоз — заболевание, характеризующееся чрезмерным прогрессирующим накоплением Fe в организме, которое откладывается в клетках паренхимы печени, поджелудочной железы и других органов. У мужчин заболевание встречается в 5-10 раз чаще, чем у женщин. Первичный (идиопатический, наследственный) гемохроматоз развивается наиболее часто (1:500) и наследуется по аутосомно-рецессивному типу.
В 80-90 % случаев определяется гомозиготная мутация Cys282Tyr в гене HFE (тип 1), что приводит к прекращению синтеза интактного гепцидина. 4-5 % пациентов с гемохроматозом гетерозиготы по мутации Cys282Tyr и одновременно гетерозиготы по мутации His63Asp гена HFE (сложные гетерозиготы). Реже гемохроматоз связан с мутацией гена самого гепцидина (тип 2А), гена HJV (тип 2В) или гена TRF2 (тип 3) либо молекулы-мишени гепцидина — транспортного белка ферропортина (тип 4).
При каждой мутации в кишечнике всасывается избыточное количество Fe, поскольку отсутствие гепцидина имитирует тяжелый дефицит Fe. Повышается концентрация сывороточного Fe, ферритина и насыщаемость трансферрина. После ранней диагностики избыток Fe (около 25-50 г по сравнению с нормой 2-5 г у здорового человека) можно нормализовать еженедельной сдачей крови в течение 1-2 лет (норма сывороточного ферритина менее 50 мкг/л, процент насыщения трансферрина менее 50 %).
Вторичный гемохроматоз наблюдается при нарушении утилизации Fe (например, повышенное всасывание с неэффективным эритропоэзом при β-талассемии или сидеробластной анемии), заболеваниях печени (например, алкогольный цирроз, портокавальное шунтирование), атрансферрине-мии, поздней кожной порфирии, а также при избыточном поступлении Fe перорально или парентерально (частые переливания крови, которые служат второй причиной нарушения использования Fe, длительный гемодиализ, инъекции препаратов Fe).
Повышенное накопление Fe (особенно в форме гемосидерина [гемосидероз]) вызываеттоксическое поражение клеток. Механизмы повреждающего действия включают:
а) опосредованное железом образование свободных радикалов (липидная пероксидация клеточных мембран);
б) повреждение ДНК;
в) повышенный синтез коллагена, инициированный железом.
После того как содержание Fe в печени превысит норму в 20 раз, развивается фиброз печени, переходящий в цирроз. Риск смерти от печеночноклеточного рака повышается в 200 раз. Сидероз вызывает фиброз поджелудочной железы и приводит к повреждению β-клеток, инсулиновой недостаточности и сахарному диабету.
Накопление меланина и гемосидерина в коже, особенно на открытых участках тела, вызывает гиперпигментацию («бронзовый диабет»). Сидероз в сердце сопровождается кардиомиопатией, аритмией и сердечной недостаточностью, приводящей к смерти в молодом возрасте.
Fe ускоряет метаболизм аскорбиновой кислоты (витамина С); дефицит витамина С способствует развитию повреждений суставов (псевдоподагра).
- Рекомендуем ознакомиться со следующей статьей "Схема развития болезни Вильсона"
Оглавление темы "Схемы патогенеза нарушений метаболизма":- Схема нарушения обмена аминокислот
- Схема нарушения обмена углеводов
- Схема нарушения обмена жиров (липидозов)
- Схема нарушения обмена липопротеидов
- Схема развития подагры
- Схема развития гемохроматоза
- Схема развития болезни Вильсона
- Схема последствий дефицита α-1-антитрипсина
- Схема развития диспротеинемий (нарушения обмена белков)
- Схема развития порфирии