
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема нарушения обмена липопротеидов
Помимо липидозов среди нарушений обмена жиров выделяют группу заболеваний, при которых в сыворотке изменяется концентрация липопротеидов и транспортлипидов. В крови липиды транспортируются в составе глобулярных молекулярных комплексов (микроэмульсии), которые называются липопротеидами.
Поверхность липопротеидов состоит в основном из амфифильных липидов (фосфолипидов и неэстерифицированного холестерина), в то время как ядро содержит неполярные (гидрофобные) липиды — триглицериды (ТГ) и эфиры холестерина (ЭХС), которые являются транспортной и резервной формой холестерина. В состав липопротеидов входят также белки, которые называются аполипопротеидами (Апо). Липопротеиды различаются по размеру, плотности (плотность определяет название класса липопротеидов, см. далее), липидномусоставу, месту синтеза, типам Апо.
Аполипопротеиды выступают в роли структурных элементов липопротеидов (например, АпоАII и Апо В48),лигандов рецепторов липопротеидов в мембранах клеток-мишеней (например, АпоВ100 и АпоЕ) и активаторов ферментов (например, AnoAl, АпоСII).
Хиломикроны транспортируют липиды из кишечника (через лимфатические сосуды кишечника) к периферическим тканям (скелетные мышцы, жировая ткань), где их АпоСII активируетлипопротеидлипазу (ЛПЛ), находящуюся в эндотелиальных клетках. ЛПЛ расщепляет ТГ хиломикронов до свободных жирных кислот (СЖК), которые поступают в клетки мышечной и жировой тканей.
Остаточные хиломикроны через АпоЕ связываются с рецепторами гепатоцитов (возможно, это белок, подобный рецептору ЛПНП) и путем эндоцитоза поступают в клетку. Таким образом, в клетки печени доставляются ТГ, холестерин и ЭХС. Поступившие в организм липиды, а также вновь синтезированные ТГ и холестерин выходят из печени и транспортируются к периферическим тканям в составе липопротеидов очень низкой плотности (ЛПОНП). АпоСН ЛПОНП в эндотелии капилляров жировой и мышечной тканей активирует ЛПЛ, что приводит к высвобождению жирных кислот.

В результате этого процесса отщепляется АпоСН, а АпоЕ экспонируется на поверхности липопротеидов. Образуются остаточные ЛПОНП или липопротеиды промежуточной плотности (ЛППП), половина из которых возвращается в печень, связываясь, в основном, через АпоЕ с рецепторами ЛПНП. В печени они «загружаются» вновь синтезированными липидами и превращаются в ЛПОНП, которые покидают печень. Вторая половина ЛППП под действием печеночной липазы превращается в липопротеиды низкой плотности (ЛПНП), при этом АпоЕ отщепляется, а АпоВ100 экспонируется на поверхности липопротеидов.
При этом 2/3 ЛПНП поставляют холестерин и ЭХС в печень, 1/3 — во внепеченочные ткани. Поступление липопротеидов в клетку осуществляется посредством связывания АпоВЮО с рецепторами ЛПНП. Взаимодействие с рецептором происходит в области покрытых клатри-ном ямок поверхности клеток. ЛПНП путем эндоцитоза поступают в клетку, а рецепторы ЛПНП снова перемещаются в клеточную мембрану (рециркуляция рецептора). После слияния эндосомы с л изосомой под действием лизосомных ферментов Апо разрушаются, ЭХС гидролизуются, свободный холестерин поступает в цитозоль. Повышение концентрации холестерина в клетке влечет за собой следующее:
1) ингибируется ключевой фермент синтеза холестерина [ГМГ-КоА-редуктаза);
2) активируется ацил-КоА-холестеринацилтрансфераза [АХАТ], катализирующая образование ЭХС, в виде которых он депонируется;
3) подавляется синтез рецепторов ЛПНП.
Липопротеиды высокой плотности (ЛПВП) обмениваются определенными Апо с хиломикронами и ЛПОНП, а также забирают избыток холестерина из внепеченочных тканей и крови. АпоАI ЛПВП активирует фермент плазмы лецитинхолестерин-ацилтрансферазу, который частично эстерифицирует холестерин. ЛПВП транспортируют холестерин и ЭХС в печень и эндокринные железы, синтезирующие стероидные гормоны (яичники, семенники, надпочечники), где имеются рецепторы ЛПВП.
Увеличение уровня липидов в сыворотке может быть обусловлено холестерином, ТГ или обоими компонентами (гиперхолестеринемия, гипертриглицеридемия или комбинированная гиперлипидемия). «Гиперлипопротеидемия» — это общий термин для обозначения различныхтипов нарушения обмена липопротеидов.
У большинства пациентов с гиперхолестеринемией концентрация холестерина в сыворотке превышает 200-220 мг/дл. Гиперхолестеринемия, причина которой остается неизвестной, часто обнаруживается у членов одной семьи (полигенная гиперхолестеринемия). Важную роль играют ожирение и особенности питания. Снижению уровня холестерина ЛПНП способствует диета с высоким содержанием растительных (ненасыщенных) жиров.
Животные (насыщенные) жиры повышают синтез холестерина в печени, уменьшают плотность рецепторов ЛПНП, что приводит к увеличению концентрации богатых холестерином ЛПНП всыворотке(холестерин ЛПВП >135 мг/мл). Это усиливает связывание Л ПНП со скэвенджер-рецепторами (рецепторы-«мусорщики»), которые опосредуют поступление холестерина в макрофаги, клетки кожи и стенок сосудов. Гиперхолестеринемия, таким образом, относится к фактору риска атеросклероза и ИБС.
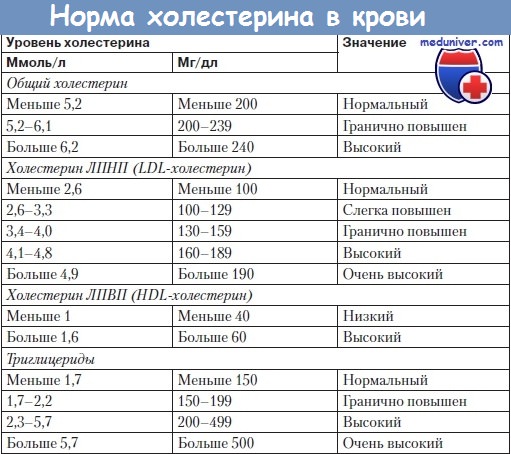
При семейной гиперхолестеринемии (гиперлипопротеидемия IIа типа, распространенность в общей популяции гомозиготной формы 1:106, гетерозиготной — 1:500) уровень холестерина плазмы значительно повышен сразу после рождения: при гомозиготной форме в 2 раза, при гетерозиготной — в 6 раз.Поэтомуинфарктмиокарда может развиваться даже у детей. Основная причина семейной гиперхолестеринемии — мутация гена, кодирующего рецептор ЛПНП, что нарушает поступление ЛПНП в клетки. Указанная мутация:
1) уменьшает активность транскрипции гена рецептора;
2) вызывает задержку белков рецептора в эндоплазматическом ретикулуме;
3) препятствует включению рецептора в клеточную мембрану;
4) уменьшает связывание ЛПНП с рецептором;
5) нарушает эндоцитоз. Повышение уровня холестерина сыворотки обусловлено двумя причинами.
Во-первых, снижается захват клетками богатых холестерином ЛПНП. Во-вторых, внепеченочные ткани синтезируют больше холестерина, т. к. при снижении поступления ЛПНП в ткани не происходит ингибирования ГМГ-КоА-редуктазы. Кроме диеты (см. выше) для лечения используют ионообменные смолы (холестирамин), которые связывают соли желчных кислот в кишечнике и прекращают их кишечно-печеночную рециркуляцию. Это повышает синтез желчных кислот из холестерина в печени и, соответственно, снижает внутриклеточную концентрацию холестерина. При гомозиготной форме увеличивается плотность рецепторов ЛПНП.
Тем не менее это также стимулирует синтез холестерина, который можно подавить с помощью ингибиторов ГМГ-КоА-редуктазы, например ловастатина. Лечение гомозиготной формы предусматривает удаление ЛПНП из плазмы с помощью плазмафереза.
При другом дефекте гена развивается комбинированная гиперлипидемия (гиперлипопротеидемия типа IIb), которая характеризуется умеренным ростом уровня ТГ и холестерина в крови. Причина — повышение продукции АпоВ, что вызывает увеличение синтеза ЛПОНП и, следовательно, образования ЛПНП. Семейная (наследственная) дис-β-липопротеидемия предрасполагает к развитию гиперлипопротеидемии типа III, при которой вместо нормального АпоЕЗ экспрессируется вариант АпоА2, который не распознается рецептором АпоЕ. Нарушается захват печенью остаточных хиломикро-нов и ЛППП, повышается их концентрация в плазме, что обусловливает высокий риск атеросклероза.
Первичная гипертриглицеридемия развивается вследствие повышенного синтеза ТГ в печени или, реже, в результате нарушения метаболизма хиломикронов и ЛПОНП (гиперлипопротеидемия типа I) из-за недостаточности ЛПЛ или АпоСII. У больных отмечается склонность к развитию панкреатита. Кроме того, снижается уровень ЛПВП, поэтому повышается риск атеросклероза (уменьшение выведения холестерина из стенки сосудов?).
Мутации генов могуттакже приводить к снижению концентрации липопротеидов в плазме (гиполипопротеидемия). Семейная гипо-а-липопротеидемия (болезнь Танжера) развивается вследствие дефекта АпоА и характеризуется уменьшением концентрации ЛПВП, что повышает риск атеросклероза. При абеталигопротеидемии в плазме отсутствуют ЛПНП (гипохолестеринемия). Заболевание обусловлено аномальным синтезом АпоВ, из-за чего хиломикроны не экспортируются из слизистой кишечника, а ЛПОНП — из печени, что вызывает накопление ТГ в обоих органах.
- Рекомендуем ознакомиться со следующей статьей "Схема развития подагры"
Оглавление темы "Схемы патогенеза нарушений метаболизма":- Схема нарушения обмена аминокислот
- Схема нарушения обмена углеводов
- Схема нарушения обмена жиров (липидозов)
- Схема нарушения обмена липопротеидов
- Схема развития подагры
- Схема развития гемохроматоза
- Схема развития болезни Вильсона
- Схема последствий дефицита α-1-антитрипсина
- Схема развития диспротеинемий (нарушения обмена белков)
- Схема развития порфирии