
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
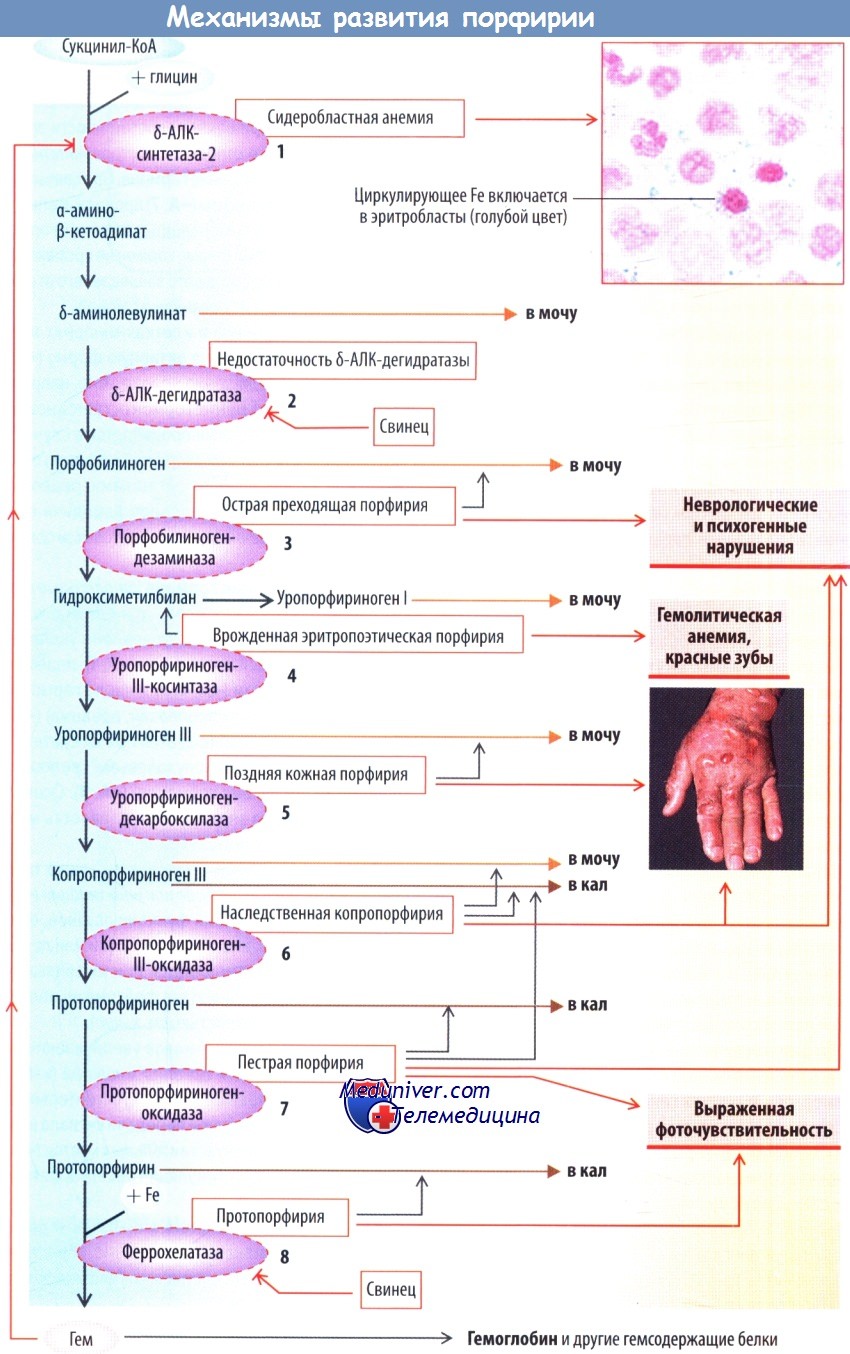
Схема развития порфирии
Синтез гема включает 8 реакций. Помимо включения в гемоглобин эритроцитов гем синтезируется практически во всех органах и входит в состав миоглобина, цитохрома Р450, каталазы, перок-сидазы и цитохромов дыхательной цепи. Поскольку гемопротеины абсолютно необходимы для организма, полное ингибирование синтеза гема несовместимо с жизнью.
Частичная (обычно у гетерозигот) недостаточность одного из ферментов, участвующих в синтезе гема, приводит к тяжелым последствиям.
Синтез гема начинается с образования α-амино-β-кетоадипата, который спонтанно превращается в δ-аминолевулинат (δ-аминолевулиновая кислота [δ-АЛК]). Этот этап происходит в митохондриях, он определяет скорость всего процесса синтеза гема и катализируется в эритробластах δ-АЛК-синтетазой-2, в печени — δ-АЛК-синтетазой-1. Активность обоих ферментов ингибируется конечным продуктом — гемом (отрицательная обратная связь).
Механизм ингибирования частично обусловлен связыванием гема в цитозоле с регуляторным элементом профермента и торможением транспорта его в митохондрии.
Проявления нарушения синтеза гема определяются механизмом обратной связи и зависят от снижения активности ферментов данного метаболического пути. При снижении активности δ-АЛК-синтетазы-2 уменьшается количество конечного продукта гема, что вызывает лишь недостаточное повышение активности этого фермента, накопление железа и развитие сидеробластной анемии.
При недостаточности ферментов следующих стадий синтеза гема по механизму обратной связи значительно возрастает количество δ-АЛК, т. к. не происходит ингибирования δ-АЛК-синтетазы. В результате концентрация субстратов всех последующих реакций увеличивается до тех пор, пока не будет синтезироваться достаточное количество тема.
Высокая концентрация промежуточных метаболитов вызывает развитие заболеваний, которые называются первичными порфириями. Растворимые в воде промежуточные метаболиты выделяются с мочой (δ-АЛК, порфобилиноген, уропорфирин), растворимые в липидах — через желчь с калом (копропорфирины, протопорфирины). Порфирины образуются из соответствующих порфиногенов. Обнаружение их в моче или кале имеет диагностическое значение.
Концентрация δ-АЛК повышается при недостаточности δ-АЛК-дегидратазы (порфобилиногенсинтазы) и порфобилиногендезаминазы (также называется гидроксиметилбилансинтетазой), что вызывает развитие острой преходящей порфирии, сопровождающейся повышением концентрации порфобилиногена. Это приводит к развитию нейровегетативных нарушений (тахикардия, тошнота, рвота, запор) и нейропсихогенных расстройств (параличи, судороги, кома, галлюцинации).
Причиной этих нарушений может быть конкуренция между δ-АЛК и нейромедиатором γ-аминомасляной кислоты, имеющей с ней структурное сходство.
При врожденной эритропоэтической порфирии из гидроксиметилбилана неферментативным путем синтезируется уропорфириноген, который под действием ферментов метаболизируется до копропорфириногена I. Копропорфириноген I не метаболизируется и выводится с мочой. У новорожденных это вызывает появление красных пятен на пеленках, а затем на зубах. К другим симптомам относятся повышенная светочувствительность кожи и гемолитическая анемия.
При поздней кожной порфирии, которая встречается чаще, порфирины вызывают поражение кожи (плохо заживающие волдыри) вследствие поглощения света, особенно с длиной волны 440 нм, и воздействия избытка свободных радикалов.
При наследственной копропорфирии, а также пестрой порфирии, особенно распространенной в Северной Африке (около 3 случаев на каждую 1000 белых жителей), повышается уровень δ-АЛК, порфобилиногена и копропорфиринов. Это вызывает нейропсихогенные и дерматологические проявления у больных детей. При протопорфирии (повышение протопорфиринов) после воздействия УФ-лучей развиваются ожоги, зуд и болезненность кожи.
Приобретенные порфирии наблюдаются при отравлении свинцом и характеризуются высоким уровнем δ-АЛК и порфобилиногена. Также они возникают при заболеваниях гепатобилиарной системы, когда снижается экскреция копрофорфирина в желчь.
- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Схемы патогенеза нарушений метаболизма":- Схема нарушения обмена аминокислот
- Схема нарушения обмена углеводов
- Схема нарушения обмена жиров (липидозов)
- Схема нарушения обмена липопротеидов
- Схема развития подагры
- Схема развития гемохроматоза
- Схема развития болезни Вильсона
- Схема последствий дефицита α-1-антитрипсина
- Схема развития диспротеинемий (нарушения обмена белков)
- Схема развития порфирии