
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Кернса—Сэйра - клиника, диагностика
Синдром Кернса—Сэйра (М1М 530000) - заболевание характеризуется развитием на 1-м или 2-м десятилетии жизни прогрессирующей наружной офтальмоплегии (птоз, ограничение движений глазных яблок, двоение) в сочетании с пигментной дегенерацией сетчатки, атриовентрикулярной блокадой сердца, миопатией, прогрессирующей мозжечковой атаксией, нейросенсорной глухотой, эндокринными расстройствами.
Мозжечковая атаксия в ряде случаев синдрома Кернса—Сэйра может быть в течение длительного времени единственным симптомом со стороны ЦНС, что связано с рано развивающейся гибелью клеток Пуркинье в коре мозжечка. При развернутой клинической картине большинство больных погибают от патологии сердца через 10—20 лет от начала заболевания.
В литературе описаны также те или иные «редуцированные» варианты синдрома Кернса—Сэйра — разновидности прогрессирующей наружной офтальмоплегии в сочетании с мышечной слабостью, снижением толерантности к физической нагрузке, умеренно выраженной атаксией (Bertini Е. et al.). На MP-томограммах у больных с синдромом Кернса—Сэйра визуализируются изменения сигнала со стороны среднего мозга (красных ядер), зубчатых ядер мозжечка и дентаторубральных волокон в верхней мозжечковой ножке, базальных ганглиев, таламуса, субкортикального белого вещества больших полушарий.
При лабораторных исследованиях обычно выявляется повышение уровня белка в цереброспинальной жидкости, лактат-ацидоз, феномен «рваных красных волокон» в мышечных биоптатах.
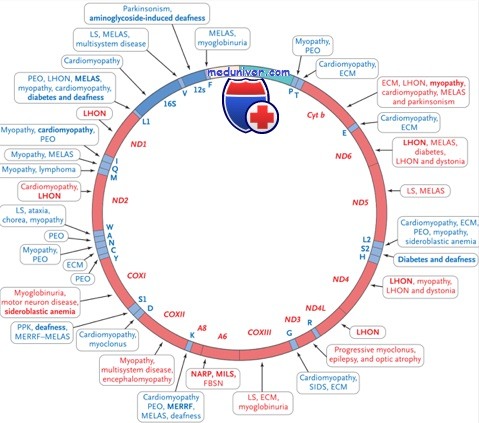
У около 80% больных с синдромом Кернса—Сэйра имеются те или иные делеции митохондриального генома протяженностью от 2 до 10,4 кб, причем 30—40% из них имеют идентичную делению 4977 нуклеотидов, захватывающую сегмент мтДНК от гена АТФазы 8 до гена ND5 (DiMauro S., Wallace D.).
Описаны более редкие случаи, когда при тех или иных вариантах синдрома Кернса—Сэйра у больных выявлялись дупликации или точковыс мутации мтДНК (DiMauro S.). Все описанные мутации являются гетероплазмическими, и различные соотношения уровней нормальной и мутантной мтДНК в разных тканях-мишенях обусловливают значительный полиморфизм клинических проявлений болезни.
В отличие от простых точковых мутаций мтДНК, характерных для MERRF, MELAS или NARP, наблюдаемые при синдроме Кернса—Сэйра делсции и иные крупные перестройки мтДНК обычно возникают de novo в соматических клетках на ранней стадии эмбриогенеза и приводят к манифестации спорадических случаев митохондриальных болезней.
Наследование этих мутаций невозможно, поскольку, как предполагается, ооциты с крупными перестройками мтДНК не способны дать начало развитию эмбриона (DiMauro S.).
- Читать "Лечение митохондриальных атаксий"
Оглавление темы "Митохондриальные атаксии":- Нейросенсорная тугоухость, атаксия и миоклонус - клиника, диагностика
- Синдром Кернса—Сэйра - клиника, диагностика
- Лечение митохондриальных атаксий
- Синдром Лея - клиника, диагностика
- Атаксии при нарушениях контроля стабильности митохондриальной ДНК
- Атаксии при нарушениях утилизации митохондриальных субстратов
- Церебротендинальный ксантоматоз (ксантоматоз мозга и сухожилий) - клиника, диагностика
- Болезнь Ниманна—Пика - клиника, диагностика
- Абеталипопротеинемия (синдром Бассена—Корнцвейга) - клиника, диагностика
- Атаксия при GM2-ганглиозидозе - клиника, диагностика