
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Кожа при синдроме Марфана - эпидемиология, этиология, клиника, лечение
Синдром Марфана - краткий обзор:
- Частота встречаемости: от 1:5000 до 1:10000 (OMIM #154700).
- Аутосомно-доминантный тип наследования с мутациями гена фибриллина 1 (FBN1; хромосома 15q21.1).
- К кожным симптомам относятся полосы растяжения кожи (стрии) (у двух третей пациентов), паховые или грыжи послеоперационного рубца и, редко, перфорирующий серпигинирующий эластоз.
- Внекожные симптомы включают переразгибание суставов, смещение хрусталика вверх, скелетные аномалии, аневризмы и разрывы аорты.
а) Эпидемиоология. Синдром Марфана является аутосомно-доминантным заболеванием, первично поражающим скелет, глаза и церебро-васкулярную систему. Около 25% случаев наблюдаются спорадически, особенно пациенты, рожденные от отцов более старшего возраста. Был описан герминативный родительский мозаицизм. Общая распространенность синдрома Марфана установлена на уровне приблизительно 1:5000-10000 человек, без расовых, половых и географических различий. За последние 30 лет прогноз для таких пациентов улучшился благодаря прогрессу в консервативном и хирургическом лечении.
б) Этиология и патогенез. Стандартное гистологическое исследование пораженной кожи не выявляет патологии, тогда как электронная микроскопия дермального коллагена при синдроме Марфана выявляет патологическое его утолщение, расположение и форму, включая фибриллы различной толщины, неупорядоченно расположенные, скрученные и фибриллы в виде цветка и с неровными краями, подобно изменениям при СЭД. Относительное содержание коллагена I к коллагену III также снижено. Недавно были описаны дефекты архитектоники коллагеновых микрофибрилл крупных сосудов, выраженные в большей степени, чем патологическое содержание коллагена или нарушение поперечных связей между фибриллами.
Конечная архитектоника сходна с той, что обнаружена в аневризмах абдоминальной части аорты, чем и объясняют слабость стенки аневризмы при синдроме Марфана.
Синдром Марфана развивается вследствие гетерозиготных мутаций в гене профибриллина 1 на хромосоме 15q21.1. Спектр клинических фенотипов, возникающих в результате дефекта FBN1, превосходит классический синдром Марфана и эти непохожие состояния названы фибриллопатиями/фибриллинопатиями, Они бывают разной степени выраженности — от тяжелого неонатального фенотипа Марфана до изолированного расширения корня аорты или марфаноидных скелетных проявлений без типичной церебро-васкулярной патологии или эктопии хрусталика. Фибриллин — это гликопротеин весом 350 кБа, который является основным компонентом экстрацеллюлярных матриксных (ЭЦМ) микрофибрилл, которые являются структурными компонентами волокон пояска поддерживающей связки хрусталика и связаны с эластичными волокнами в аорте и коже.
Теоретически каждая семейная мутация индивидуальна и было описано около 500 мутаций в каком-либо из 65 экзонов гена. Не было выявлено «горячих точек», за исключением случаев неонатального синдрома Марфана. Интересно отметить, что мутации, вызывающие преждевременную терминацию кодона, приводят к более легкой форме синдрома, а мутации в экзонах 24-31 связаны с неонатальным синдромом Марфана и более тяжелыми проявлениями заболевания. Синдром Марфана II типа вызван мутациями в локусе гена TGFBR2.
Фибриллин взаимодействует с латентным TGF-β-связывающим протеином, блокируя его и контролируя активность TGF-β. Недостаточность фибриллина повышает конститутивную (клеточную) активность TGF-β, и мутация фибриллина-1 приводит к активации латентного TGF-β, с действием через «классический» и неклассический пути. В сосудистых тканях TGF-β считается основным медиатором структуры и функции сосудистого экстрацеллюлярного матрикса (ЭЦМ), посредством его депонирования и разрушения в матриксе. Повышенное действие TGF-β усиливает деградацию ЭЦМ, что приводит к развитию и росту аневризмы. Стимуляция любого из действий TGF-β также может приводить к фосфорилированию Smad, который включает профибротический сигнальный ответ. Ангиотензин II действует через рецепторы двух типов, типа 1 (АТ 1) и типа 2 (АТ 2), которые оказывают противоположные действия.
Ангиотензин II, воздействуя через АТ 1, также повышает экспрессию лигандов TGF-β и рецепторов и стимулирует фиброз сосудистой стенки. Дополнительно к действию активированного TGF-β, макроагрегаты мономеров фибриллина образуют основные каркасные структуры, на которых собираются зрелые волокна эластина; исходя из этого, патологически измененный фибриллин образует беспорядочный матрикс из микрофибрилл, который может в дальнейшем приводить к измененной и слабой формации эластических волокон и разрыву сети микрофибрилл, которые соединяют эластическую пластинку и соседние интерстициальные клетки.
В разработке находятся исследования по влиянию на эти патогенетические пути и их потенциальный терапевтический эффект при синдроме Марфана. Лозартан, блокатор рецепторов 1 типа к ангиотензину II, также выдал обещающие результаты путем замедления дилатации корня аорты у пациентов с синдромом Марфана. Лозартан не только блокирует TGF-β, но как оказывается, также уменьшает фосфорилирование Smad2 независимо от его TGF-β эффектов. Таким образом, блокада АТ1 может прерывать несколько патологических путей, которые вызывают ремоделирование сосудистой стенки и прогрессию заболевания.

Обратите внимание на высокий рост, арахнодактилию, ненормально низкое соотношение верхнего и нижнего сегментов тела, длинные руки.
в) Клиника. Синдром Марфана является системным заболеванием соединительной ткани, проявляющимся патологическими изменениями в трех системах органов: глаза (обычно вывих хрусталика); скелет (избыточная длина конечностей, разболтанные суставы, деформации передней стенки грудной клетки и кифосколиоз); и наиболее важные церебро-васкулярные изменения (обычно аневризма аорты и пролапс митрального клапана). Многие из типичных проявлений синдрома Марфана зависят от возраста, что затрудняет постановку диагноза в детстве. Ни один клинический симптом не является патогномоничным.
Для «марфаноидного хабитуса» характерен долихостеномелический (высокий и худой) тип телосложения, с более вытянутым нижним сегментом туловища (лобковый симфиз направлен в пол), который длиннее верхнего сегмента (рост минус нижний сегмент). Характерно, что длина размаха рук превышает рост человека на несколько сантиметров. Дистальные кости имеют избыточную длину (арахнодактилия). Скелетные проявления синдрома Марфана характеризуются избыточным ростом костей и гипермобильностью суставов. Кифосколиоз может быть выраженным и нарастать по мере взросления. Патология грудной клетки, например, pectus excavatum (воронкообразная деформация грудной клетки, впалая грудь) или килевидная грудь (выпячивание грудины), являются результатом избыточного роста ребер и встречаются часто. Комбинация кифосколиоза и воронкообразной деформации грудной клетки иногда вызывают нарушение функции легких и сердца. Гипермобильность суставов из-за разболтанности капсулы, связок и сухожилий может вызвать плоскостопие, гиперподвижность коленных и локтевых суставов (genu recurvatum) и иногда вывих сустава.
Вывих надколенника не является редкостью; вывих бедра, часто выявляемый в период новорожденности, может быть первым признаком синдрома Марфана. Скрининговые тесты на гипермобильность суставов — это симптом большого пальца (или симптом Стейнберга), при котором большой палец легко сгибается и укладывается поперек ладони, и симптом запястья (Walker-Murdoch), при котором концевая фаланга большого пальца перекрывает концевую фалангу мизинца при охвате ими запястья противоположной руки. Гипермобильность суставов и удлиненные конечности у пациентов с синдромом Марфана часто позволяют им обернуть руки вокруг спины и достать пупок с противоположной стороны.
У большинства пациентов с синдромом Марфана имеется миопия в связи с уплощением роговицы и патологически длинной переднезадней осью орбиты. Установлено, что у 50%-70% пациентов имеется эктопия хрусталика, обычно в виде верхнего вывиха хрусталика. Подвывих и полный вывих хрусталика часто приводят к вторичным патологиям глаза, включая аметропию, миопию, острую глаукому и повышенные риск отслойки сетчатки. Поскольку легкий подвывих можно пропустить при стандартном офтальмологическом осмотре, то при подозрении на синдром Марфана необходим осмотр с помощью щелевой лампы.
Церебро-васкулярные изменения обусловливают большинство случаев заболеваемости и смертности у больных синдромом Марфана. Церебро-васкулярные изменения выявлены у 40% пациентов при исследовании сердца и почти у 100% пациентов при посмертном обследовании. Медиальный некроз аорты является самой частой патологией, и, как правило, происходит диффузная дилатация проксимального сегмента восходящей аорты с аортальной регургитацией. Такая дилатация нарастает и может даже определяться в брюшной полости. Нарастание дилатации происходит непостоянно, и эта непредсказуемость требует частого мониторирования. Смерть у пациентов с синдромом Марфана обычно случается во взрослом возрасте в результате цереброваскулярных осложнений, чаще всего вторично на фоне дилатации корня оарты, которая приводит к расслоению стенки аорты (или разрыву) и тампонаде сердца. Пролапс митрального клапана (ПМК) возникает в результате расширения митрального кольца, с растяжением хорд и избыточным движением створок митрального клапана.
Это происходит примерно у 25% пациентов детского и подросткового возраста, и у 86% имеющих впалую грудную клетку. ПМК усиливается с возрастом и впоследствии наблюдается у 75% пациентов. ПМК может привести к патологическим изменениям на электрокардиограмме, появлению митральной регургитации и даже сердечных аритмий, приводящих к внезапной смерти.
Самыми частыми кожными проявлениями синдрома Марфана являются недостаточность подкожно-жировой клетчатки, наличие стрий, наиболее заметных на верхней части грудной клетки, руках, бедрах и животе. Эти кожные проявления обнаруживаются почти у двух третей пациентов. У пациентов с синдромом Марфана часто наблюдается ползучий перфорирующий эластоз, и могут развиваться паховые и послеоперационные грыжи. Кожные проявления считаются малыми диагностическими признаками в пересмотренных диагностических критериях.
Дуральная эктазия (расширение твердой мозговой оболочки в пояснично-крестцовом отделе) часто развивается у пациентов с синдромом Марфана. Была описана эмфизема, легочные буллы повышают риск пневмоторакса, особенно поражая верхние доли легких. В ротовой полости к симптомам данного заболевания относятся готическое небо и скученное положение передних зубов.
г) Неонатальная форма синдрома Марфана. Новорожденные с неонатальной формой синдрома Марфана имеют диспропорциональное тело, дряблую кожу, эмфизему, патологию глаз, контрактуры суставов, кифосколиоз, приведенные бедра, морщинистые уши, микрогнатию, мышечную гипоплазию и недостаточность подкожно-жировой клетчатки вокруг суставов. Тяжелая недостаточность сердечного клапана и аортальная дилатация приводят к смерти в течение первых двух лет жизни.
д) Синдром Марфана у детей. В недавнем ретроспективном исследовании выявили клинические проявления у детей в группе более чем 1000 пациентов с мутациями FBN1. Средний возраст при постановке диагноза—6,5 лет, и только у 30% был положительный семейный анамнез. Пациенты с неонатальной формой синдрома Марфана составляли 14% когорты. Из больных детей у 19% была тяжелая форма заболевания, у 32% классический синдром Марфана и у 35% пациентов — подозрение на синдром Марфана. В популяции детей у 71% уже обнаруживалась дилатация восходящего отдела аорты. Эктопия хрусталика наблюдалась примерно у половины детей, типичные марфаноидные проявления в скелете—у 28%. Расширение дуги аорты чаще было у новорожденных, тем не менее, аортальные осложнения были редкостью среди детей. В неясных случаях выявление мутаций FBN1 часто облегчало диагностику.
В то время как неонатальная форма синдрома Марфана проявляется как тяжелая ранняя форма de novo, сопровождаемая высокой смертностью, пациенты, прошедшие диагностику после подросткового возраста, обычно имеют сходный со взрослыми пациентами фенотип. В более легких случаях чаще встречался положительный семейный анамнез, скорее всего это было вызвано тем, что в более легких случаях пациенты чаще доживали до репродуктивного возраста. Важно, что у больных детей при первом осмотре зачастую не определяются интернациональные критерии синдрома Марфана. Поскольку симптомы нарастают с возрастом, наблюдение детей при подозрении на данную патологию является решающим для постановки диагноза.
и) Лаборатоные исследования. Диагноз синдрома Марфана основан на совокупности клинических симптомов. Патогномоничного диагностического лабораторного теста или четких гистологических изменений не существует. Повышение гидрокси-пролина и десмозина в моче также не патогномонично. Офтальмологический осмотр опытным специалистом и, при наличии показаний, проведение операции должны осуществляться как можно раньше. Ортопедический осмотр, при показаниях, и эхокардиографический мониторинг размеров корня аорты и функции клапанов сердца являются важнейшими, как и кардиологическое пособие, оказываемое в связи с найденными изменениями.
к) Дифференциальный диагноз. Синдром Марфана иногда ошибочно можно принять за такое заболевание, как, например, гомоцистинурию. Тем не менее, ряд патологий вызывает фенотип, частично пересекающийся с проявлениями синдрома Марфана, включая некоторые, обусловленные мутациями фибриллина-1. Эти данные суммированы в блоке ниже.
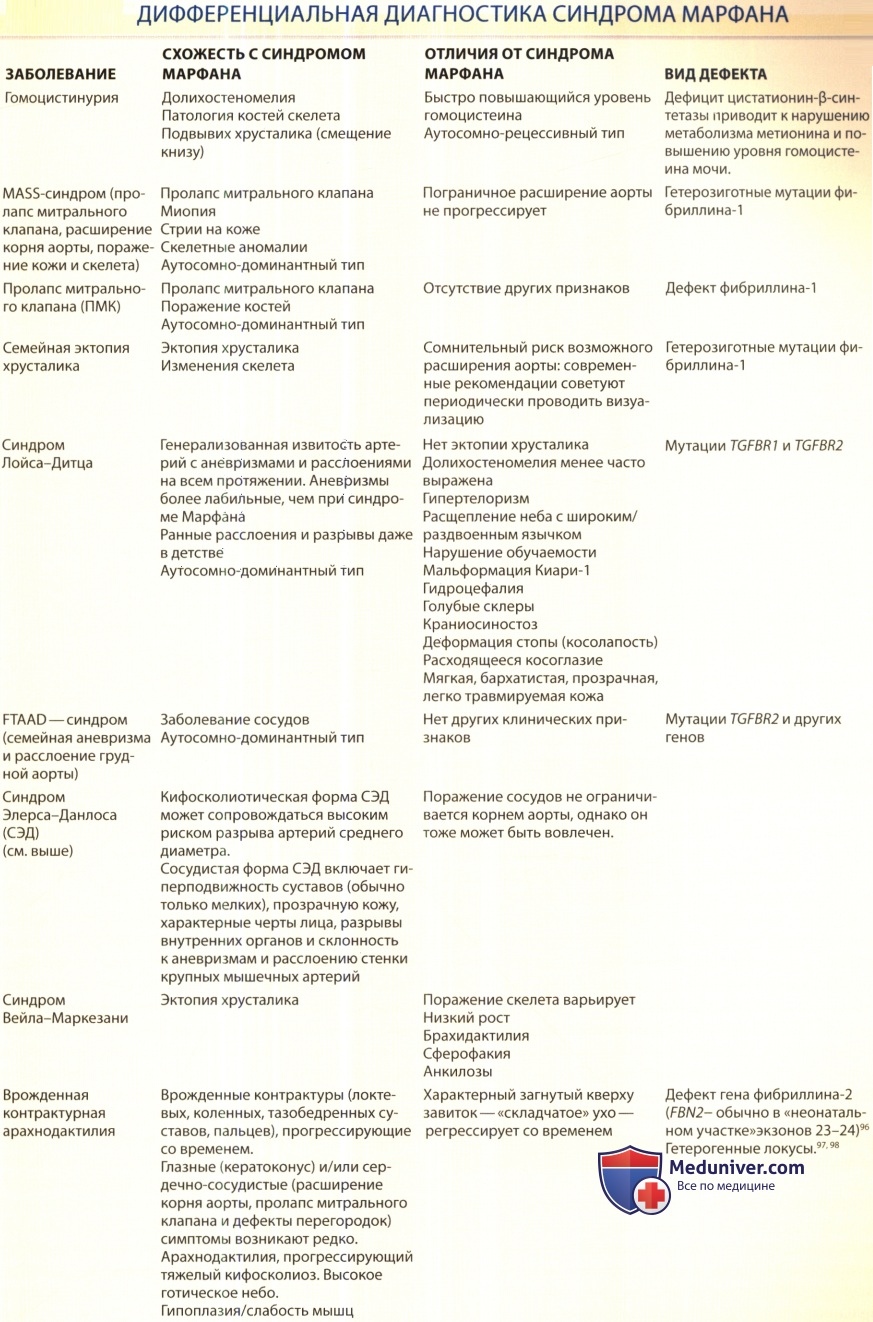
л) Лечение синдрома Марфана. Лечение синдрома Марфана сфокусировано на профилактике инвалидности и угрожающих жизни потенциальных осложнений. Ранние и регулярные офтальмологические осмотры необходимы для выявления излечимой амблиопии и отслойки сетчатки. Эктопия хрусталика и даже полный вывих могут сохраняться десятилетиями. Экстракция хрусталика бывает необходима для лечения диплопии, глаукомы, катаракты или отслойки сетчатки. Лазерная хирургия противопоказана.
При нарушении функции сердечно-легочной системы проводят лечение воронкообразной деформации грудной клетки, но только после полного созревания скелета, чтобы предотвратить рецидив деформации. В дальнейшем будет необходимо удалить внутренние стабилизирующие конструкции. Сколиоз может уменьшиться у девочек-подростков на фоне эстрогенной терапии, но это может вызвать задержку роста. Бандажи, физиотерапия и стабилизация позвоночника бывают необходимы для профилактики тяжелого сколиоза.
Наличие в семейном анамнезе ранней расслаивающей аневризмы аорты требует тщательнейшего мониторинга. Пациенты с синдромом Марфана должны, как минимум, ежегодно проходить мониторинг. Аортальные осложнения у пациентов с нормальным диаметром аорты, сохраняющимся в течение нескольких лет, выявлены не были (у взрослых). Длительное лечение пропранололом рекомендовано для профилактики расширения аорты путем снижения сократительной функции миокарда, но он не влияет на выживаемость. У небольшой группы детей с синдромом Марфана, не поддающегося лечению β-блокаторами, блокаторы ангиотензина II типа 1 (лозартан и ирбесартан) вызывают значительное уменьшение диаметра корня аорты и расширения синотубулярного соединения.
Продолжаются более крупные рандомизированные исследования по влиянию этих препаратов. Патологические изменения клапанов сердца и аневризматические дефекты могут потребовать протезирования, но такую операцию следует отложить, насколько возможно, чтобы избежать повторной замены протеза, особенно у растущих детей. Операция на корне аорты приводит к увеличению средней продолжительности жизни и показана, если максимальный размер более 5 см у взрослых и старших детей, скорость увеличения размера около 1 см/год или при прогрессивном развитии аортальной регургитации. Пациентам с выявленным расширением аорты следует избегать приема кофеина, стрессовых ситуаций и силовых упражнений. Пациенты с поражением легких должны избегать ситуаций с быстрым изменением атмосферного давления, например, подводного плавания с аквалангом или авиаполетов, и, разумеется, не курить.
Доксициклин ингибирует матриксную металлопротеиназу и, как было показано, улучшает архитектонику аортальной стенки и отсрочивает расслоение аорты в модели мышей. Будущие исследования могут сфокусироваться на синергии между доксициклином и лозартаном, поскольку матриксные металлопротеиназы могут активировать TGF-β.
Дети освобождаются от занятий физкультурой, чтобы избежать потенциально вредных нагрузок, контактных видов спорта и изометрических упражнений, которые могут привести к разрыву аорты или к сердечной недостаточности. К сожалению, всё это лишь усиливает изоляцию ребенка, который уже может беспокоиться о том, как выглядит его необычное тело, или является социально отверженным (изгоем) из-за того, что выглядит «другим» или слишком высоким. Важно, что не все пациенты с синдромом Марфана имеют «классический» фенотип. Если у пациентов с атипичной внешностью подозревают данный диагноз, нужно быть уверенным, что проведены все соответствующие исследования для того, чтобы не пропустить потенциально опасные внутренние изменения.
- Рекомендуем далее ознакомиться со статьей "Эластичная псевдоксантома (синдром Гренблада-Странберга) - этиология, гистология, лечение"
Редактор: Искандер Милевски. Дата публикации: 14.12.2018