
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Кожа при врожденном дискератозе (синдром Цинссера-Коула-Энгмана)
Врожденный дискератоз, или синдром Цинссера-Коула-Энгмана — Х-сцепленное полисистемное заболевание с поражением кожи, слизистых оболочек, глаз, желудочно-кишечного тракта, а также гематологическими нарушениями и повышенной частотой развития раковых опухолей. Также существуют как аутосомно-доминантные, так и аутосомно-рецессивные формы.
В базу данных одной из лондонских клиник было включено 228 семей с 354 больными из 40 стран. В литературе было описано более 500 случаев. У больных женщин может развиться либо аутосомно-доминантная, либо аутосомно-рецессивная форма заболевания.
а) Клиника. К наиболее типичным признакам относится триада симптомов, включающая ретикулярную гиперпигментацию, дистрофию ногтей и лейкоплакию слизистых оболочек. В течение первых десяти лет жизни у пациентов на участках кожи, подверженных солнечному воздействию, развивается ретикулярная пойкилодермия в сочетании с гиперпигментацией и в редких случаях образованием пузырей. Дистрофия ногтей отмечается практически у всех пациентов, начиная с двух до пяти лет.
Изначально ногти слегка расслаиваются, затем образуются продольные гребни с неправильными свободными краями. В конечном итоге ногти уменьшаются в размере, что приводит к остаточным рудиментам. Ногти на пальцах рук поражаются обычно до поражения ногтей на пальцах ног.
К другим кожным симптомам относятся атрофичность и сморщенность кожи на тыльной поверхности кистей и стоп, а также гипергидроз и гиперкератоз ладоней и ступней с исчезновением кожных борозд (отсутствие отпечатков пальцев).
Лейкоплакия может поражать слизистую оболочку любой локализации. Слизистая оболочка полости рта поражается наиболее часто, но лейкоплакия также может обнаруживаться в уретре, на головке полового члена, во влагалище или прямой кишке.
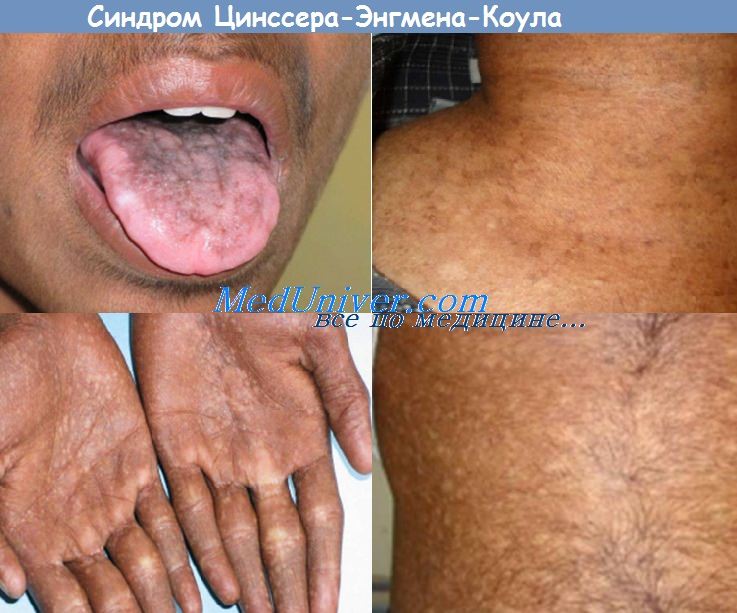
Может развиваться гиперкератоз языка. Слизистые оболочки пищевода, уретры и слезного протока могут сокращаться и стенозироваться, что приводит к дисфагии, дизурии и ретенционному слезотечению соответственно. Часто встречаются множественный кариес зубов и их раннее выпадение.
Прогрессирующее поражение легких, в том числе инфекции и фиброз (и даже цирроз у некоторых пациентов), было выявлено у 20% пациентов мужского пола. Получено несколько сообщений о внутричерепных кальцификатах, особенно в базальных ганглиях. Последние также наблюдаются при синдроме Коккейна (СК).
Отмечается повышенная частота развития новообразований, особенно плоскоклеточного рака полости рта, прямой кишки, шейки матки, влагалища, пищевода и кожи. У крупного родового клана Британии была выявлена болезнь Ходжкина и аденокарцинома поджелудочной железы.
Как и при анемии Фанкони (АФ), хотя гипоплазия является основным патологическим процессом, наблюдаемым в костном мозге, при данном заболевании отмечается предрасположенность к миелодисплазии и острому миелоидному лейкозу.
У большинства пациентов (93%) развивается аплазия костного мозга, которая служит основной причиной ранней смерти (у 71% пациентов). Примерно у половины пациентов (21-41%) развивается панцитопения с гематологической картиной, сходной с таковой при анемии Фанкони (АФ), в возрасте до 10 лет. Некоторым пациентам была выполнена трансплантация костного мозга.
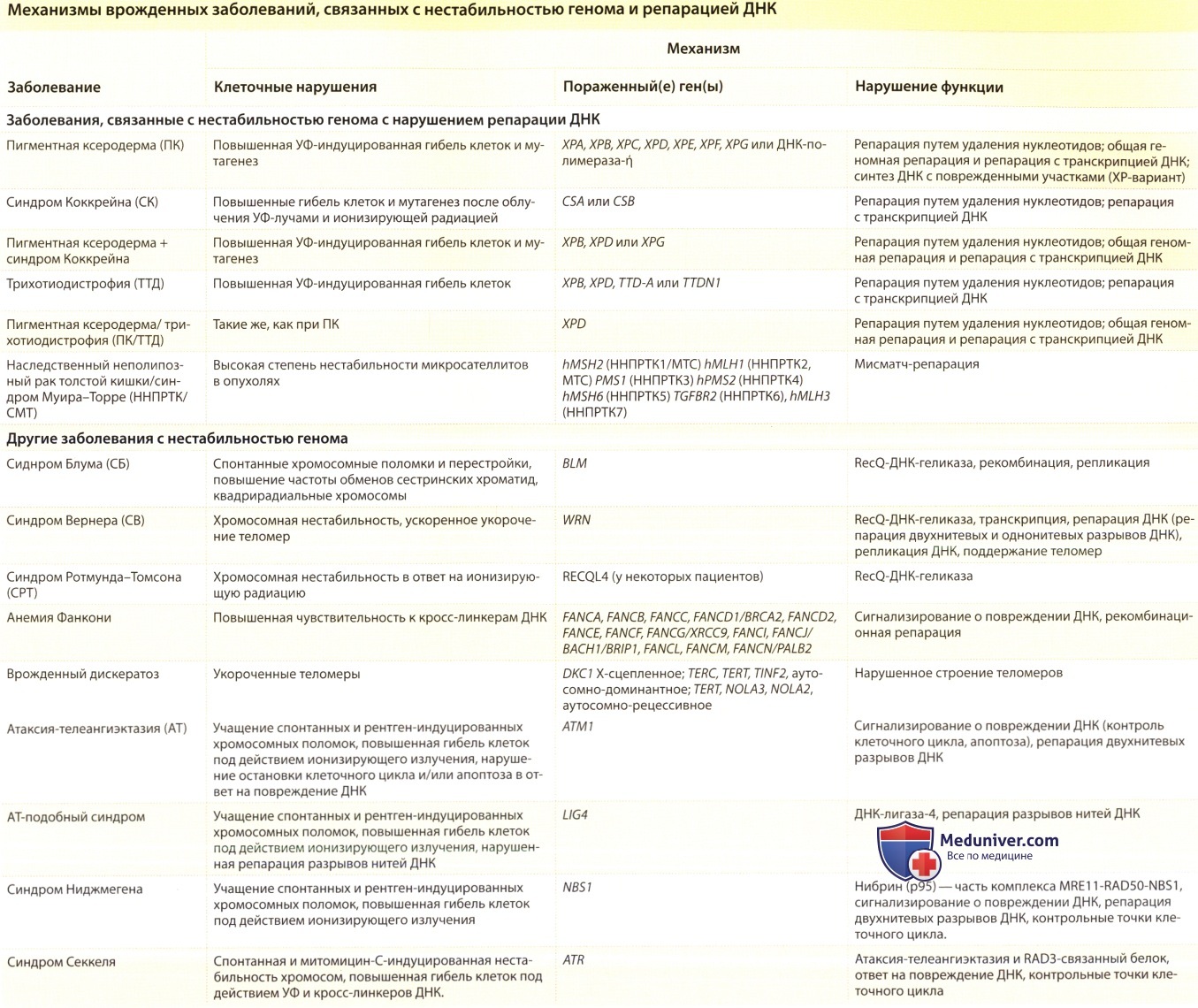
б) Анализы при врожденном дискератозе. Был клонирован ген врожденного дискератоза (DKC1), локализованный на участке q28 Х-хромосомы. Белок дискерин имеет ядерную функцию и играет роль в сохранении теломер и процессе старения. У больных были обнаружены мутации в гене DKC1. Т
акже получены данные указывающие на аутосомную форму заболевания у женщин с мутациями гена TERC, который также вовлечен в функцию теломер.
Примерно у половины пациентов с врожденным дискерацитозом имеется мутация в гене в теломере биологического пути: DKCl, TERC, TERT, TINF2, NOLA2, and NOLA3. У многих пациентов с БД очень короткие теломеры.
- Рекомендуем далее ознакомиться со статьей "Причины и механизмы развития туберозного склероза"
Редактор: Искандер Милевски. Дата публикации: 22.12.2018