
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Сегментарный нейрофиброматоз. Синдром Нунан. Синдром Легиуса. Шванноматоз
а) Сегментарный нейрофиброматоз 1 типа. Термин «сегментарный нейрофиброматоз» подразумевает признаки НФ-1 (обычно пятна «кофе с молоком» и нейрофибромы), ограниченные одной областью тела. Ruggieri и Huson предложили для таких случаев термин «мозаичный локализованный НФ-1» в качестве подтверждения того, что патогенез этого заболевания заключается в развитии мутации гена НФ-1 после зачатия, что ведет к соматическому мозаицизму.
Это было подтверждено у пациента, у которого мутантный аллель NF-1 находился в мозаичном виде в культивированных фибробластах из пятен «кофе с молоком» и отсутствовал в фибробластах нормальной кожи.
Преобладающее большинство пациентов с сегментарным НФ-1 имеют пятна «кофе с молоком» или пигментные пятна по типу веснушек в области кожных складок и только в одной области тела. В таких случаях имеется риск развития осложнений НФ-1 в пораженной области, чаще всего — нейрофибром. Локализованные признаки НФ-1, помимо пятен «кофе с молоком», пигментных пятен или кожных нейрофибром также могут представлять собой примеры соматического мозаицизма.
В нескольких работах сообщалось о наличии изолированных плексиформных нейрофибром, в том числе — в одном случае, при котором отмечалась утрата гетерозиготности гена НФ-1 в шванновских клетках опухоли. Так как лишь 50% случаев псевдоартроза большеберцовой кости развивается при подверженном НФ-1, другие 50% могут на самом деле представлять собой случаи сегментарного НФ-1. Другие примеры сегментарного НФ-1 включают случай односторонней глиомы зрительного нерва у ребенка, которая биологически проявляла себя как НФ-1 ассоциированная опухоль, а также клинические случаи с изолированной дисплазией клиновидной кости.
Генетическое консультирование пациентов с сегментарным НФ-1 затруднено. Многим из таких пациентов неверно ставился диагноз НФ-1, что приводило к беспричинному беспокойству и неадекватному генетическому консультированию. Кроме того, при НФ-1 было показано существование полового мозаицизма. У пациентов с сегментарным НФ-1 рождались дети с полным НФ-1.
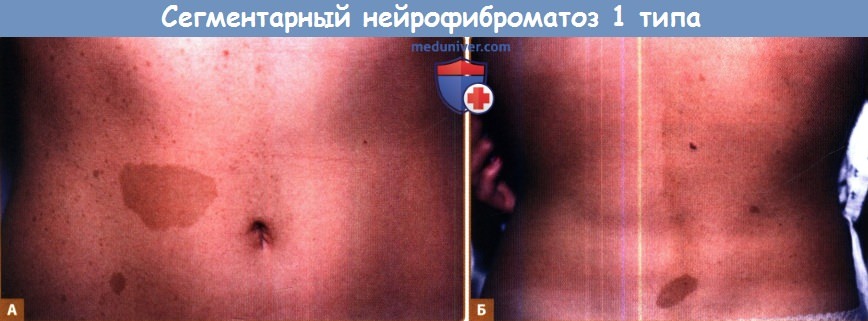
Обратите внимание на пятна цвета кофе с молоком и пигментные пятна по типу веснушек, ограниченные одной стороной тела.
б) Синдром Нунан (нейрофиброматоз 1 типа). Было установлено, что в некоторых случаях, пациенты, отвечающие диагностическим критериям НФ-1, имеют признаки синдрома Нунан, характеризующегося гипертелоризмом, птозом, антимонголоидным разрезом глаз, низко посаженными и развернутыми кзади ушными раковинами, перепончатой шеей, деформациями грудной клетки и низкорослостью. Более 50% детей с синдромом Нунан имеют поражение сердечно-сосудистой системы, наиболее часто—стеноз легочного клапана. Синдром Нунан вызван мутацией в гене PTPN11 в 50% случаев.
Также была обнаружена связь ряда других мутаций с данным фенотипом (KRAS, SOS1, В RAF, МЕК1, МЕК2, HRAS и RAF1). Генетическое исследование выявило, что у большинства пациентов с синдромом LEOPARD также определяются мутации этого гена. Недавнее исследование выявило наличие мутации в НФ-1 гене у 16 из 17 несвязанных между собой пациентов с клиническими проявлениями НФ-1 -Нунан синдрома, при отсутствии мутации в гене PTPN11. Таким образом, по-видимому, преобладающее число случаев НФ-1-Нунан связаны с мутациями гена NF-1.
в) Синдром Легиуса. Недавно были описаны семейные случаи аутосомно-доминантного наследования пятен «кофе-с-молоком», опрелых веснушек и макроцефалии при отсутствии других признаков НФ-1, включая нейрофибромы, при которых не была обнаружена мутация НФ-1. Дальнейшие генетические исследования этих пациентов показали наличие мутаций в SPRED1, гене, отрицательно влияющем на синтез митоген-активированной протеинкиназы (МАПК).
Ни у одного из пациентов не были обнаружены очаговые нейрофибромы, плексиформные нейрофибромы, узелки Пиша, опухоли зрительных нервов или костные деформации, характерные для НФ-1. Все еще неизвестна частота встречаемости когнитивных нарушений в данных случаях. Генетическое тестирование на мутации в SPRED1 доступно для пациентов с подозрением на синдром Легиуса, при котором не обнаруживаются мутации в гене НФ-1.
г) Шванноматоз. Некоторое время назад было отмечено, что у отдельных пациентов развиваются множественные шванномы, но нет других признаков НФ-2, в частности, вестибулярных шванном. У пациентов с данным состоянием, которое теперь называется шванноматозом, развиваются множественные болезненные шванномы периферических нервов и параспинальных нервных корешков. Опухоли чаще всего начинают появляться на втором и третьем десятилетии жизни. Для купирования постоянных болей, как правило, требуется хирургическое удаление отдельных опухолей.
В отличие от пациентов с НФ-2, пациенты с шванноматозом имеют нормальную продолжительность жизни. До выставления диагноза шванноматоза крайне важно исключить возможность НФ-2 посредством генетического анализа и проведения МРТ для выявления вестибулярных шванном. Хотя в большинстве случаев шванноматоз развивается спорадически, была зарегистрирована аутосомно-доминантная передача этого синдрома. Мутации зародышевой линии в SMARCВ1/IN11, другом гене опухолевой супрессии, ранее рассматривались как ответственные за образование рабдоидных опухолей у младенцев и маленьких детей, но оказались также причиной развития большого количества случаев семейного шванноматоза.
- Вернуться в оглавление раздела "дерматология."
Редактор: Искандер Милевски. Дата публикации: 22.12.2018