
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Схема развития нарушений почечного транспорта
Поражение канальцевых транспортных процессов может быть обусловлено генетическими, токсическими факторами, лекарственными препаратами или гормональными изменениями.
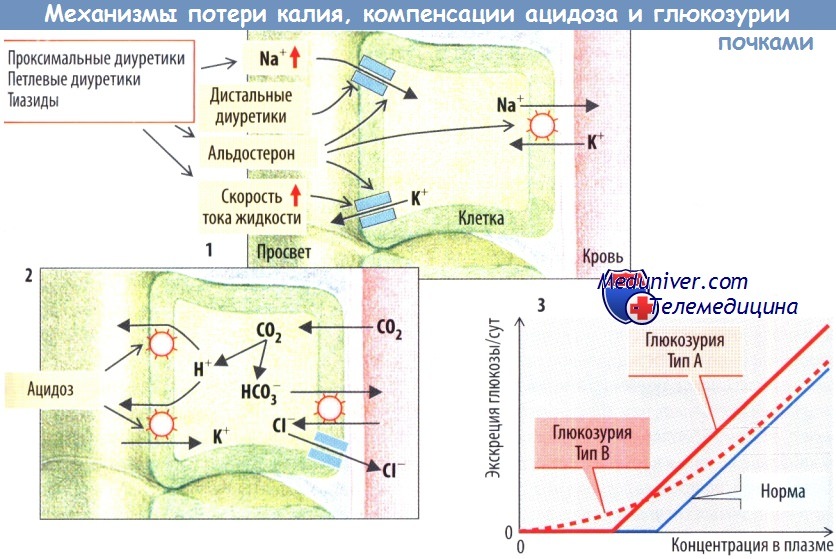
За реабсорбцию глюкозы в проксимальных канальцах отвечает как минимум два мембранных переносчика. Генетический дефект почечного или кишечного Na+-глюкозо/галактозного переносчика SGLT1 (SLC5A1 приводит к глюкозо-галактозной мальабсорбции и лишь к незначительной глюкозурии. Повреждение второго почечного переносчика глюкозы SGLT2 (SLC5A2) сопровождается классической почечной глюкозурией, при которой снижена либо максимальная скорость переноса (тип А), либо аффинность (тип В). Тип 0 характеризуется полным отсутствием резорбции глюкозы. Глюкоза выходит из клетки через базолатеральную клеточную мембрану с помощью универсальных переносчиков GLUT2 (SLC2A2). Генетический дефект этого транспортера приводит к глюкозурии, гипер- и гипогликемии, депонированию гликогена и рахиту (синдром Фанкони—Биккеля).
Фактор роста фибробластов 23 (FGF23) ингибирует Na+-фосфатный котранспортер. Этому способствует и почечный гормон Klotho. Как правило, FGF23 расщепляется протеазой РНЕХ (фосфатрегулирующий гомолог эндопептидазы на Х-хромосоме). Генетические повреждения способны инактивировать РНЕХ (Х-сцепленный гипофосфатемический рахит) либо к образованию РНЕХ-резистентного FGF23 (аутосомно-доминантный гипофосфатемический рахит). В обоих случаях транспорт фосфатов в почечных канальцах ослаблен (почечный фосфатный диабет). Клетки некоторых опухолей продуцируют FGF23 и дополнительные фосфотонины, которые ингибируют фосфатный транспорт. Почечная реабсорбция фосфатов, кроме того, нарушается при дефиците кальцитриола.
Потеря почками фосфатов служит причиной деминерализации костей вследствие дефицита фосфатов (рахит). Повышенная почечная реабсорбция фосфатов, например, при дефиците ПТГ (гипопаратиреоз) или нарушенной активности ПТГ (псевдогипопаратиреоз) ведет к гиперфосфатемии.
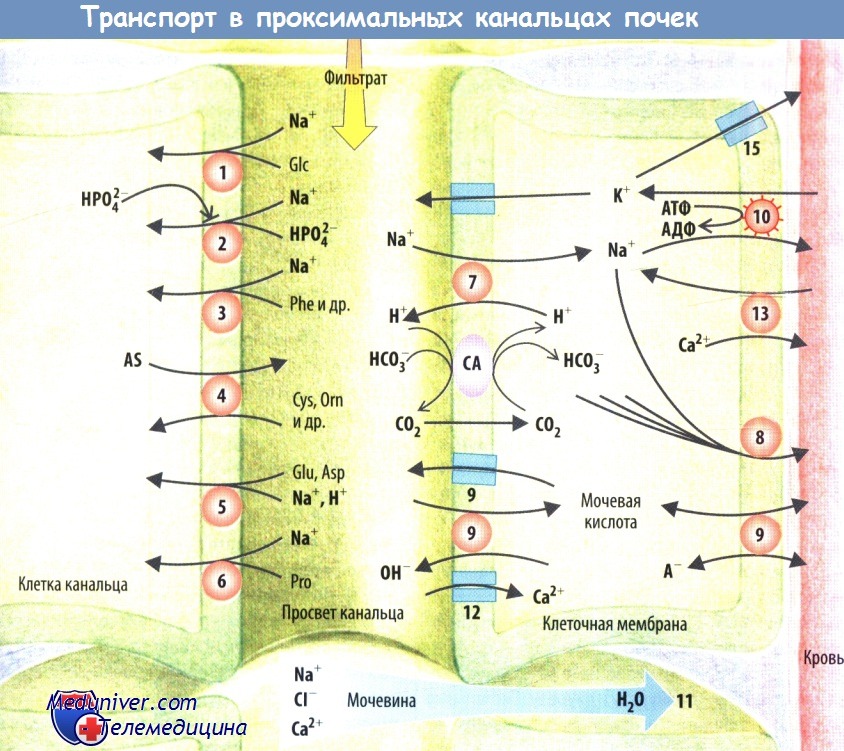
Нарушение Na+-котранспорта некоторых нейтральных аминокислот (В0АТ1 [SLC6A19]) в почках и кишечнике приводит к болезни Хартнупа, при которой наблюдается повышенная экскреция аминокислот. Потеря почками триптофана снижает синтез никотиновой кислоты, вызывая ее дефицит и, следовательно, поражение нервной системы и кожи.
Дефект переносчика нейтральных и двухосновных аминокислот (В0+АТ-rВАТ) усиливает выведение почками орнитина, лизина и цистина (цистинурия). Плохо растворимый цистин осаждается, образуя мочевые камни. При семейном нарушении белковой толерантности наблюдается аномальная реабсорбция двухосновных аминокислот (Y+LAT1-4F2hc [SLC3A2/SLC7A7]).
Повреждение Na+-котранспортера кислых аминокислот (ЕААТЗ [SLC1A1]) сопровождается неопасной для человека аминоацидурией; дефект переносчика циклических аминокислот, таких как пролин,- иминоглицинурией.
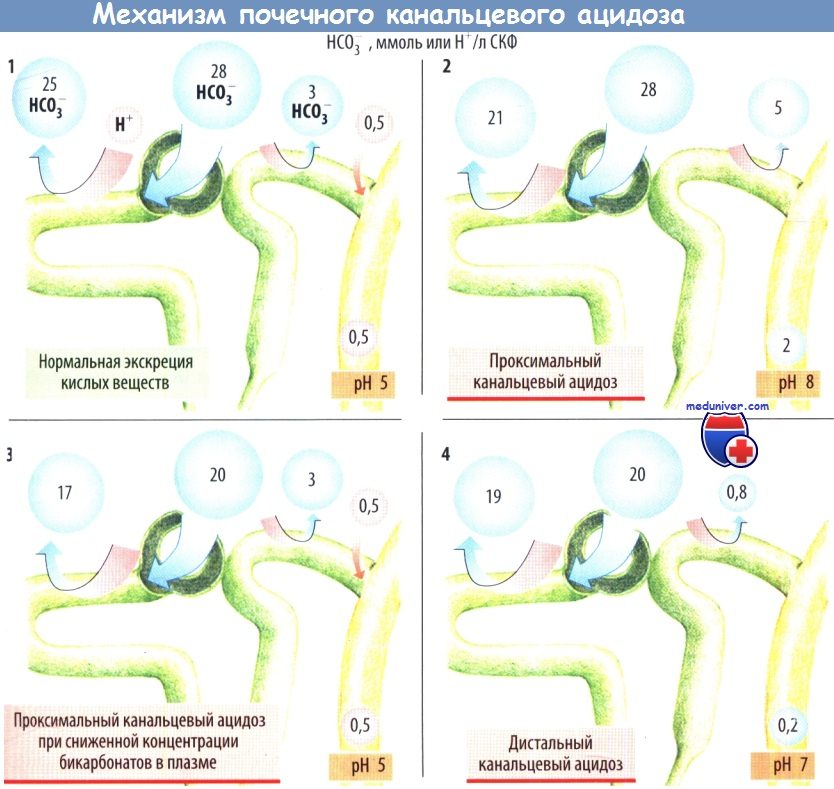
Пониженная активность Na+/H+-обменника NHE3 или Na+-HCO3--котранспортера NaCl приводит к ацидозу в проксимальных канальцах. Поскольку уменьшение реабсорбции HCO3- в проксимальных канальцах не может быть компенсировано слабым транспортом в дистальных канальцах, бикарбонаты выделяются с мочой, даже если нагрузка HCO3- находится в пределах нормы.
Если концентрация HCO3- в плазме снижается, проксимальные канальцы способны реабсорбировать почти все количество отфильтровавшихся бикарбонатов; в результате в дистальных канальцах образуется моча с нормальной кислотностью. Повреждение карбоан-гидразы (СА) уменьшает секрецию H+ в проксимальных и дистальных канальцах (тип III RTA). Na+-HCO3- котранспорт зависит от мембранного потенциала. Гиперкалиемия вызывает деполяризацию клеточной мембраны и угнетает реабсорбцию HCO3- в проксимальных канальцах, в то время как при гипокалие-мии реабсорбция HCO3- увеличивается. Таким образом, почечная экскреция H+ зависит от концентрации внеклеточного K+.
Уменьшение объема межклеточной жидкости стимулирует Na+/H+-обменник и, соответственно, реабсорбцию HCO3- в проксимальных канальцах. Это приводит к алкалозу вследствие уменьшения объема межклеточной жидкости. Угнетение активности Na+/H+-обменника либо карбоангидразы усиливает выведение натрия (натрийурия). Ослабление реабсорбции Na* в проксимальных канальцах в значительной степени компенсируется усилением его реабсорбции в дистальных сегментах нефрона (в петле Генле).
При синдроме Фанкони, обусловленном генетическими или приобретенными (например, интоксикация) факторами, нарушаются некоторые Na+-связанные процессы транспорта, что приводит к глюкозурии, аминоацидурии, фосфатурии, ацидозу в проксимальных канальцах, гипокалиемии.
Вследствие увеличения реабсорбции Na+ и воды в просвете канальцев растет концентрация мочевой кислоты, за счет чего повышается ее реабсорбция через люминальные и базолатеральные анионные обменники и каналы. Это вызывает гиперурикемию и накопление плохо растворимой мочевой кислоты в некоторых суставах (подагра).
Дефицит энергии (например, недостаточное кровоснабжение) сопровождается снижением активности Na+/K+-АТФазы, что ведет к уменьшению реабсорбции электролитов (нефропатия с потерей солей), набуханию и гибели клеток.
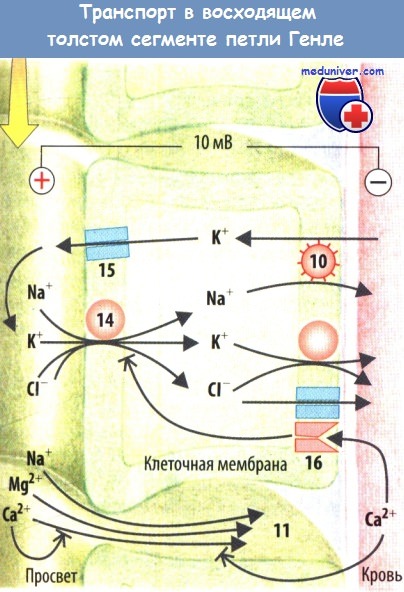
Реабсорбция Са2+ в проксимальных канальцах, петле Генле и дистальных канальцах происходит частично путем околоклеточного транспорта, через Са2+-каналы в апикальной мембране и с помощью 3Na+/Са2+-обменников перитубу-лярной мембраны. ПТГ стимулирует реабсорбцию Са2+; гипопаратиреоз, напротив, приводит к гиперкальциурии. Высокая концентрация Са2* (гиперкальциемия) подавляется путем стимуляции Са2+-рецептора, Na+-K+-2Cl--котранспортера (NKCC2), за счет околоклеточных шунтов. Это снижает реабсорбцию не только Са2+, но и Mg2+ (потеря магния) и Na+ (натрийурия, нарушение концентрирующей функции почек).
Угнетение NKCC2 петлевыми диуретиками останавливает реабсорбцию NaCl и, соответственно, концентрирование мочи. Это приводит к массивному натрийурезу и диурезу. Дистальные канальцы и собирательные трубочки переполнены Na+, а также реабсорбируют Na+ взамен на К+, что сопровождается калийурией и гипокалиемией. NKCC2 нуждается в К+ как в субстрате, который должен рециркулировать через К+-каналы (внешние медуллярные К+-каналы, ROMK). При дефиците К+ или гипокалиемии К+-канал закрыт, реабсорбция NaCI в петле Генле ослаблена.
Генетический дефект NKCC2, Cl- или К+-каналов — причина синдрома Бартера, при котором нарушается концентрирование мочи, наблюдается натрийурия, гипокалиемия (за исключением дефектов ROMK), снижается АД, несмотря на повышенную активность ренин-ангиотензин-альдо-стероновой системы. Компенсаторноеувеличе-ние реабсорбции Na+ в дистальных канальцах притормаживается простагландинами, которые образуются вследствиеувеличения нагрузки на транспортную систему. Ингибирование ЦОГ препятствует критическому снижению объема межклеточной жидкости.
Реабсорбция NaCl в петле Генле уменьшается при гиперкальциемии, например, путем блокировки околоклеточных шунтов и активации Са2+-рецепторов.
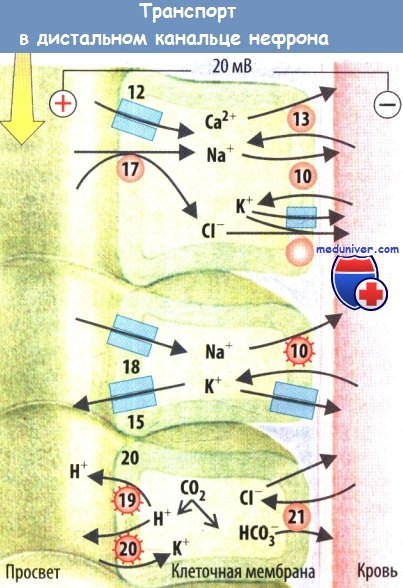
NaCl реабсорбируется в начале дистальных канальцев путем Na+-Cl-котранспорта. Тиазиды служат причиной потери натрия и калия из-за угнетения переносчиков. Уменьшение объема межклеточной жидкости повышает реабсорбцию Na+ и Са2+, что вызывает антикальциурию. Генетический дефект переносчика проявляется синдромом Гительмана, легкий вариант синдрома Бартера. Наследственный дефект WNK-киназ обычно ведет к угнетению переносчиков и развитию гипертензии (синдром Гордона).
Na+ реабсорбируется в конце дистальных канальцев и собирательных трубочках через апикальные Na+-каналы и базолатеральную Na+/K+-АТФазу. Приток Na+ деполяризует клетки апикальной мембраны и стимулирует секрецию K+ через апикальные K+-каналы. Если реабсорбция Na+ в проксимальных канальцах, петле Генле или в начале дистальных канальцев угнетена, большее количество Na+ достигает конечных дистальных отделов нефрона и реабсорбируется там в обмен на K+.
В результате почки теряют K+. Альдостерон активирует Na+-каналы и Na+/K+-АТФазу. Дефицит альдостерона (гипоальдостеронизм) или снижение его эффективности (псевдогипоальдостеронизм, например, из-за поврежденных Na+-каналов) приводит к почечной потере Na+, уменьшению объема внеклеточной жидкости и снижению АД. Дистальные диуретики блокируют рецепторы альдостерона (антагонисты альдостерона) или полностью ингибируют Na+-каналы. Эти препараты вызывают незначительную натрийурию и задержку почками K+. Гиперактивность Na+-каналов (синдром Лиддла), наоборот, сопровождается задержкой Na* и гипертензией.
H+-АТФаза, H+/К+-АТФаза и анионный обменник АЕ1 активируют секрецию H+ в конечных отделах дистальных канальцев и собирательных трубочках. Нарушение активации приводит к ацидозу в дистальных почечных канальцах RTA1. У людей с такими нарушениями выделяется лишь умеренно кислая моча даже при низкой концентрации HCO3- в плазме. У этих лиц в щелочной моче быстро осаждаются фосфаты, что приводит к образованию камней из СаНРO4.
Вода может реабсорбироваться во всех частях нефрона, за исключением восходящей петли Генле. Однако реабсорбция воды в дистальных канальцах и собирательных трубочках зависит от АДГ. Отсутствие АДГ или снижение чувствительности нефрона к АДГ вызывает несахарный диабет.
- Рекомендуем ознакомиться со следующей статьей "Схема нарушения концентрации мочи почками"
Оглавление темы "Патогенез болезней в схемах":- Схема регуляции почечной экскреции и ее нарушения
- Схема развития нарушений почечного транспорта
- Схема нарушения концентрации мочи почками
- Схема развития поликистоза почек
- Схема развития гломерулонефрита
- Схема развития нефротического синдрома
- Схема развития пиелонефрита (интерстициального нефрита)
- Схема развития острой почечной недостаточности (ОПН)
- Схема развития хронической почечной недостаточности (ХПН)
- Схема повышения давления при болезни почек