
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Патофизиология острого тубулярного некроза. Механизмы развития
Истинное поражение почек, приводящее к ОПН может быть классифицировано в зависимости от первичной области повреждения почечной паренхимы на повреждение клубочков, интерстициальный нефрит, васкулопатию и острый тубулярный некроз. Острое поражение клубочков может быть результатом воздействия препаратов или инфекций, интерстициальный нефрит может отмечаться при лекарственной аллергии, также может развиваться сосудистое повреждение; описанные причины в редких случаях являются первичными этиологическими факторами ОПН у пациентов с травмой. Значительно чаще отмечается повреждение канальцев, в частности клеток восходящей части петли Генле, расположенной во внешней части мозгового вещества почек. Данные клетки особенно чувствительны к гипоксии и воздействию нефротоксинов, и их повреждение является центральным звеном патофизиологии острого тубулярного некроза.
Разработан ряд животных моделей для исследования патофизиологии ишемической и токсической почечной недостаточности. Двусторонняя перевязка почечной артерии или введения в почечную артерию норадреналина с последующей реперфузией имитировали клиническую картину ишемического/реперфузионного повреждения. Введение контрастных веществ, индометацина и другие методы моделирования позволяют исследовать развитие токсического острого тубулярного некроза. В недавних исследованиях также использовались клеточные культуры, изолированные канальцы и изолированные кровоснабжаемые почки. По результатам экспериментальных и клинических исследований было уточнено несколько патофизиологических механизмов, характерных для различных экспериментальных моделей и клинических ситуаций.
Очаговая ишемия. Объединяющей темой данных исследований является баланс поступления кислорода к почечным канальцам (очаговый кровоток) и потребление кислорода (работа клеток почечных канальцев в качестве насоса). Почечный кровоток направлен непосредственно к коре для оптимизации клубочковой фильтрации. С другой стороны, кровоток в почечном мозговом веществе должен оставаться низким для сохранения осмотического градиента и усиления концентрации мочи путем конкурентного обмена. Кислород диффундирует из артериальных в венозные прямые сосуды, что приводит к относительной гипоксии наружного мозгового вещества. В медуллярном толстом восходящем колене петли Генле создается осмотический градиент за счет активной реабсорбции натрия, протекающей с большим потреблением кислорода.
Медуллярное парциальное давление кислорода во внешней части мозгового вещества в норме варьирует от 10 до 20 мм рт. ст.
Ряд защитных механизмов направлен на предотвращение внешнего мозгового вещества от повреждения вследствие нарушения кислородного баланса. На клетках внешнего мозгового вещества расположены рецепторы медиаторов, управляющих медуллярным кровотоком и гомеостазом кислорода. К вазодилататорам относятся простагландин Е 2, оксид азота и уродилантин. К вазоконстрикторам относятся ангиотензин и эндотелии. Аденозин, высвобождаемый из аденозинтрифосфата (АТФ) во время окислительного стресса является вазодилататором в большинстве систем. В почках аденозин индуцирует вазоконстрикцию в коре и вазодилатацию в мозговом веществе, что предполагает его роль в ослаблении гипоксии мозгового вещества. Несмотря на направленность описанных механизмов на сохранение внутрипочечного кровотока, тяжелые ишемические повреждения или непосредственное воздействие токсинов на канальцы может превысить компенсаторные возможности.
В таком случае некоторые из упомянутых медиаторов могут инициировать и усиливать повреждение почечных канальцев.
Недавно было продемонстрировано нарушение микрососудистых защитных механизмов при ишемическом/реперфузионном повреждении. Ранние сосудистые изменения включают формирование внутриклеточных молекул адгезии эндотелия (ICAM-1), адгезию и активацию полиморфноклеточных нейтрофилов, формирование фактора активации тромбоцитов (PAF) и усиление продукции сильных вазоконстрикторов эндотелиального происхождения, таких как эндотелин-1.
В стойкой фазе ОТН сосудистые изменения характеризуются повышенной базальной резистентностью сосудов, повышенной проницаемостью сосудов, повышенной чувствительностью к вазоконстрикторам и снижением или отсутствием чувствительности к вазодилататорам эндотелиального происхождения. Другие факторы, влияющие на постишемическую вазоконстрикцию включают повышение уровня ангиотензина II, повышение концентрации кальция в цитоплазме гладкомышечных клеток и снижение уровня тромбоксана А2. Снижение почечного кровотока сохраняется при постишемическом ОТН, адгезия нейтрофилов и тромбоцитов приводит к дальнейшей закупорке капилляров.
Таким образом, медиаторы, поддерживающие нормальный почечный кровоток, могут напротив приводить к продолжающейся ишемии с утратой нормальных механизмов обратной связи. Кроме инициирования повреждения почек, данные факторы могут создавать предрасположенность к дальнейшему повреждению почек при последующих эпизодах гипотензии, воздействии токсических препаратов или вазоконстрикторов.
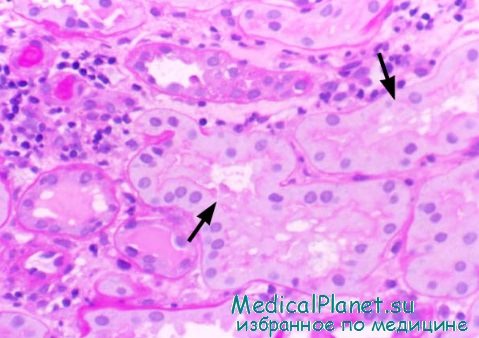
Повреждение клеток почечных канальцев
Морфологические изменения клеток канальцев после ишемического повреждения включают утрату щеточковой каймы, полярности и целостности плотных соединении проксимальных клеток проксимальных канальцев. Отмечается перераспределение Na/K-АТФазы с базальной поверхности со снижением эффективности транспорта натрия. Мертвые или гибнущие клетки попадают в просвет канальца и способствуют его закупорке. Непроходимые участки канальцев приводят к увеличению внутриканальцевого давления и снижению скорости клубочковой фильтрации. Утрата эпителиальных клеток и разрывы плотных соединений между жизнеспособными клетками приводят к обнажению базальной мембраны и обратному току клубочкового фильтрата в интерстициальное пространство.
Ишемическо-реперфузионое повреждение вызывает стремительный выброс активных метаболитов кислорода. Источники формирования окислителей в почках включают системы ксантиноксидазы, нейтрофилы и циклокосигеназы. Активные формы кислорода участвуют в повреждении почек за счет разрушения белковых структур и перекисного окисления липидов в клеточных мембранах. Результаты некоторых исследований на животных подтверждают возможность применения антиоксидантов или поглотителей активных форм кислорода при экспериментальной почечной недостаточности, в то время как в ходе других исследований подтверждение этого факта не получено.
Ишемия приводит к истощению запасов АТФ с высвобождением аденозина, инозина и гипоксантина, что приводит к вазоконстрикции. Истощение запасов АТФ активирует клеточные фосфолипазы, повышает продукцию метаболитов арахидоновой кислоты и деградацию фосфолипидов, что дестабилизирует клеточные мембраны. В ходе некоторых исследований на крысах было предположено, что экзогенное введение АТФ и магнезии защищает от ишемического повреждения почек.
Истощение запасов АТФ в клетках и дестабилизация клеточных мембран также приводит к повышению концентрации кальция в цитоплазме канальцевых и эпителиальных клеток. Внутриклеточный кальций усиливает способность митохондрий к секвестрации, высокий уровень кальция приводит к отеку митохондрий (изменению проницаемости), разобщению окислительного фосфорилирования, активации кальций зависимых протеаз и фосфолипаз, разрывам мембраны и цитоскелета и в конечном счете к гибели клетки. Кроме того, повышение концентрации кальция в цитоплазме гладкомышечных клеток приводит к вазоконстрикции и дальнейшему усилению ишемии. Фосфолипаза А2 (PLA2) активируется после ишемии-реперфузии и нарушает проницаемость клеточных и митохондриальных мембран.
Более того, PLA2 усиливает продукцию метаболитов арахидоновой кислоты, медиаторов со способностью к вазоконстрикции и хемотаксису нейтрофилов. Недавно была рассмотрена двойственная роль оксида азота в патогенезе ишемической ОПН (Goligorsky и Rabelink).
Острая ишемия почек индуцирует повышение экспрессирования индуцируемой синтазы оксида азота; блокирование фермента олигонуклеотидами, блокирующими чувствительность к стимуляции, обеспечивает функциональную защиту (по крайней мере, у крыс). Поглощение оксида азота приводит к образованию пероксинитрита, являющегося причиной повреждения канальцев во время ишемии.
Воспаление, манифестирующее как инфильтрация и активация нейтрофилов, активация системы комплимента и высвобождение цитокинов, играет роль во всех моделях истинной почечной недостаточности. Более того, воспалительные медиаторы, высвобождаемые эндотелием после ишемии-реперфузии почек могут привести к опосредованному макрофагами увеличению проницаемости легочных сосудов. Такое увеличение проницаемости легочных сосудов может быть устранено путем ингибирования активации макрофагов, что не прекращает течение ОПН.
Адгезия нейтрофилов к сосудистому эндотелию опосредована молекулами ICAM-1, расположенными на эндотелиальных клетках, связывающих рецепторы CDlla/CD18, расположенные на нейтрофилах. Синтез и проявление молекул внутриклеточной адгезии повышается в течение нескольких минут после ишемии. После адгезии и активации нейтрофилы высвобождают активные формы кислорода, протеазы, эластазу и миелопероксидазу. Взаимодействие между полиморфноклеточными нейтрофилами и эндотелием может привести к эндотелиальной дисфункции и отеку, продлевающему ишемию.
При моделировании почечной, миокардиальной, интерстициальной ишемии, истощение нейтрофилов или блокада молекул адгезии нейтрофилов защищают от ишемического-реперфузионного повреждения. Продемонстрировано также, что система комплимента также ингибирует восстановление при ОПН (в ходе исследований на животных) за счет удлинения или усиления активации лейкоцитов. Цитокины и хемокины, образующиеся местно (в ответ на клеточные стресс-факторы) или системно (как часть иммунного ответа), могут увеличивать почечную вазоконстрикцию или сокращение мезангиальных клеток, усиливая таким образом ишемию мозгового вещества и снижая фильтрационное давление.
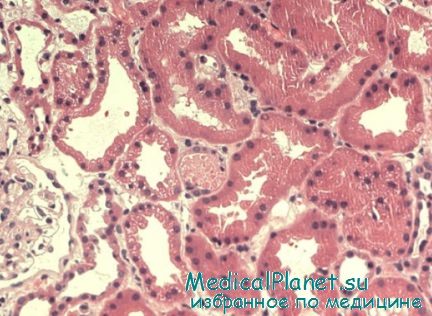
В ходе недавних исследований моделей сепсиса у животных было продемонстрировано, что транскрипция хемокинов и адгезия нейтрофилов и моноцитов к эндотелию почечных сосудов связаны с развитием ОПН. Развитие анурии и удаление азота мочевины крови и креатинина в сочетании с заметным усилением выработки хемокинов семейства СС и СХС при данной модели перитонита предполагает, что воспаление само по себе играет роль в ОПН вне зависимости от механизма ишемии-реперфузии.
После ишемии-реперфузии или токсического повреждения клетки почечных канальцев подвергаются следующим процессам: апоптозу (преимущественно клетки дистального сегмента нефрона), некрозу (преимущественно клетки проксимальных прямых и проксимальных извитых канальцев) или дифференцировке и пролиферации, приводящей к восстановлению функции канальцев. В настоящее время исследования сосредоточены на изучении внутриклеточных процессов, влияющих на описанные исходы, что позволяет надеяться на возможность направить клетки по пути пролиферации и восстановления вместо некроза и апоптоза.
В некоторых случаях внутриклеточные молекулярные процессы, отмеченные при клеточном стрессе, напоминают изменения, выявляемые при воздействии на клетку различных факторов роста (например, стремительное, краткосрочное экспрессирование немедленно-ранних (IE) генов). Продукты IE генов включают факторы транскрипции c-fos и c-jun и такие цитокины как интерлейкин-10. Активация c-jun транскрипции зависит от фосфорилирования стрессактивированными протеинкиназами (SAPK).
Активация SAPK во время IE ответа обычно оказывает антипролиферативное действие. SAPK активируются оксидативным или гипертоническим стрессом, а также фактором некроза опухолей-альфа и липополисахаридами. Кроме того, оксидативный стресс активирует ингибиторы циклин-зависимой киназы (CDK), которые блокируют нормальный клеточный цикл и приводят к апоптозу. С другой стороны, активация гена IE факторами роста опосредована внеклеточными регулирующими киназами (ERK), которые приводят к пролиферации клетки. Антиоксидант N-ацетилцистеин (NAC) блокирует ингибиторы SAPK и CDK, которые обычно активируются оксидативным стрессом. В ходе эксперимента, введение NAC до или сразу после ишемии почек ингибировало активацию гена IE и усиливало восстановление почек.
В исследованиях на моделях ишемии-реперфузии почек также была продемонстрирована активация эндонуклеаз с фрагментацией ДНК и апоптозом. Считается, что активация эндонуклеаз является ранним событием, как минимум частично являющимся причиной гибели клеток после ишемии-реперфузии, независимо от вида клеточной гибели (апоптоза или некроза). Активация протоонкогенов, таких как с-тус, отмечается в клетках почечных канальцев после воздействия различных экспериментальных стресс-факторов и, как известно, увеличивает чувствительность клеток к действию эндонуклеаз и апоптозу. Продемонстрировано, что bcl-2, преимущественно за счет противоапоптозных механизмов, в экспериментальных условиях оказывает защитное действие на клетки почечных канальцев при ОПН.
Тем не менее, точная роль протоонкогенов и эндонуклеаз в активации острого тубулярного некроза остается неясной. Так как понятны различия молекулярного ответа на ишемию-реперфузию и токсическое действие, данные пути могут дать возможность предотвращать повреждение клеток или усиливать восстановление клеток.
В отличие от большинства органов почки могут полностью восстанавливать нормальную структуру и функции после повреждения. Продуцируемые в почках факторы роста играют роль в пролиферации, могут ингибировать апо-птоз и инициировать или активировать биосинтез липидов и белков. Факторы роста, продуцируемые в ответ на острое повреждение, включают гепарин-связывающий эпидермальный фактор роста (HB-EGF), фактор роста гепатоцитов (HGF), инсулиноподобный фактор роста-1 (IGF-1) и трансформирующий фактор роста-бета (TGF-b).
Другой потенциальной ролью факторов роста после повреждения почек является повышение регионального кровотока, что было продемонстрировано экспериментально при применении IGF-1. Эпидермальный фактор роста, фактор роста гепатоцитов и IGF-1 при введении животным, подвергающимся ишемии почек, снижают выраженность нарушения функции почек и усиливают восстановление почек. Экзогенный IGF-1 у крыс усиливал восстановление почек после ишемии-реперфузии за счет усиления пролиферации канальцевого эпителия, увеличения почечного кровотока и скорости клубочковой фильтрации. Недавно было продемонстрировано, что IGF-1 защищает пациентов от почечной недостаточности после хирургических вмешательств на супраренальном сегменте аорты.
- Читать далее "Восстановление после повреждения почек - ОПН"
Оглавление темы "Острая почечная недостаточность при травме - ОПН":- Частота острой почечной недостаточности (ОПН). Факторы риска
- Физиология почек. Функции
- Патофизиология острой почечной недостаточности (ОПН). Механизмы развития
- Патофизиология острого тубулярного некроза. Механизмы развития
- Восстановление после повреждения почек - ОПН
- Синдром острой почечной недостаточности. Острая нефропатия
- Аминогликозиды как причина ОПН. Механизмы
- Амфотерицин В как причина ОПН. Механизмы
- Нефропатия вызванная контрастными веществами. Механизмы
- Нефропатия вызванная пигментами. Механизмы