
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Аутосомно-доминантные болезни - синдром Марфана
В геноме каждого человека не менее 50 тысяч пар структурных генов, локализованных в 44 аутосомах (22 пары) и в паре половых хромосом (XY — у мальчиков, XX — у девочек). Каждый ген представлен двумя аллелями (отцовским и материнским), за исключением генов в половых хромосомах мальчиков.
Описано более 4.000 наследуемых заболеваний (дефекты одного гена — моногенные болезни) с различным типом наследования (аутосомный доминантный — SR, аутосомный рецессивный — р, X/Y — сцепленный с Х-хромосомой, сцепленный с Y-хромосомой).
Аутосомно-доминантные синдромы
Общая характеристика:
a. Тип поражения. Аутосомно-доминантные синдромы обычно (хотя и не всегда) — результат мутации гена, кодирующего конкретный белок.
b. Риск передачи. Больной с аутосомно-доминантным заболеванием в 50% случаев передаёт мутантный ген потомству. Таким образом, каждый ребёнок при одном больном родителе имеет 50% риск унаследовать заболевание.
c. Тип наследования. Обычно мутантный ген наследуется от одного из родителей. Но иногда это может быть первым случаем проявления аутосомно-доминантного заболевания в данной семье — новая мутация, произошедшая в яйцеклетке или сперматозоиде.
d. Клиника:
(1) Индивидуальная вариабельность проявления — характерный признак экспрессии мутантного гена. Например, поликистоз почек у одних больных проявляется ранней почечной недостаточностью, в то время как у других и в том же возрасте — только гипертензией при сохранении нормальной функции почек.
Тяжесть клинических проявлений аутосомно-доминантных синдромов у потомков, как правило, не зависит от тяжести заболевания у родителей.
(2) Плейотропность. Мутантный доминантный ген обычно оказывает влияние на несколько тканей или систем органов.
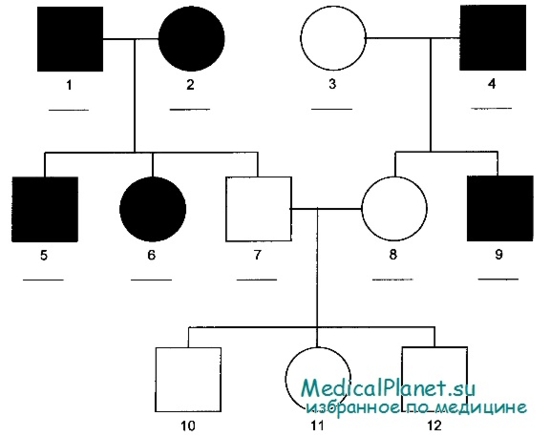
Синдром Марфана
Синдром Марфана встречается с частотой 1 на 20.000 новорождённых. Для него характерна вариабельная экспрессивность, плеиотропность и высокая популяционная частота новых мутаций.
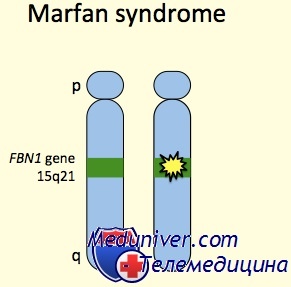
Тип поражения. Считается, что возникновение синдрома Марфана обусловлено мутацией в гене фибриллина, одного из структурных белков основного вещества соединительной ткани, что приводит к различным аномалиям её развития.
При синдроме Марфана обнаружены нарушения обмена кислых мукополисахаридов (гликозаминогликанов) типа хондроитинсерной и гиалуроновой кислот как в волокнах, так и в основном веществе соединительной ткани, что приводит к избыточному накоплению гликозаминогликанов в организме и выделению их с мочой. Также нарушается обмен оксипролина — существенного компонента коллагена. Указанные биохимические отклонения могут служить основанием для подтверждения синдрома Марфана и дифференциальной диагностики с клинически сходным заболеванием аминокислотного обмена — гомоцистинурией. b Клиника
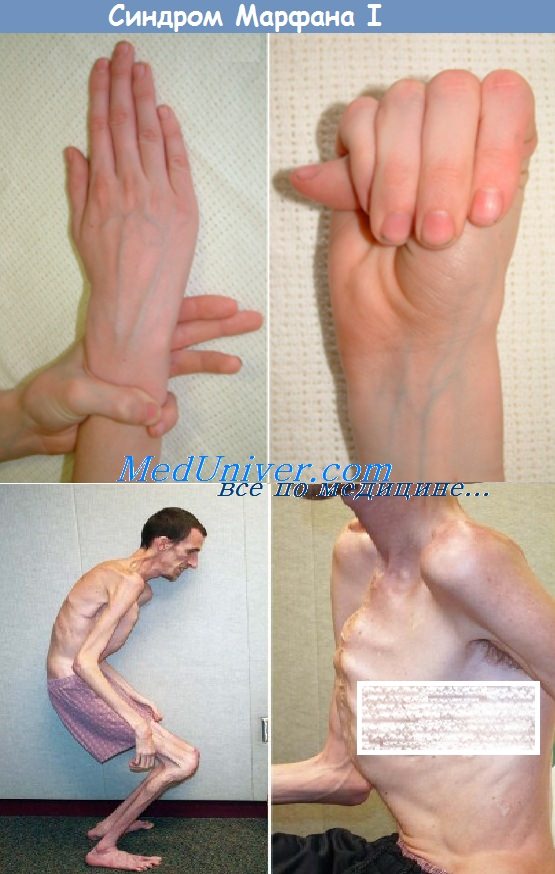
(1) Аномалии развития скелета придают больному характерный внешний вид:
(a) Необычно длинные и тонкие пальцы (арахнодактилия) и конечности (долихос-теномелия), диспропорционально высокий рост.
(b) Сколиоз и деформация грудной клетки (килевидная и/или воронковидная форма).
(c) Повышенная подвижность (разболтанность) суставов.
(d) Длинное тонкое лицо с готическим нёбом.
(2) Изменения глаз:
(a) Миопия (обычно тяжёлая)
(b) Возможен подвывих хрусталика.
(3) Кардиоваскулярные нарушения — наиболее тяжёлые проявления синдрома Марфана.
(a) Дилатация устья аорты приводит к аортальной недостаточности, а за счёт некроза средней оболочки аорты в любое время может произойти расслоение стенки и разрыв аорты. Динамический контроль за диаметром устья аорты на фоне лечения р-адреноблокаторами для предотвращения дальнейшей дилатации и превентивная хирургическая операция по замещению проксимальной части аорты заметно снижают летальность.
(b) Очень часто наблюдается пролапс митрального клапана.
(4) Лёгочные нарушения в детском возрасте менее выражены, чем у взрослых: часто наблюдается спонтанный пневмоторакс за счёт разрыва лёгочных «булл» и эмфизема лёгких.
Диагноз синдрома Марфана ставят на основании комплекса чётких клинических признаков.
(1) Диагностические критерии:
(a) Для постановки диагноза необходимо не менее двух из следующих признаков:
(I) Типичные изменения скелета
(II) Типичные глазные проявления
(III) Типичные кардиоваскулярные проявления
(IV) Отягощенный семейный анамнез
(b) Для постановки диагноза необходимо провести измерения скелета, офтальмологическое исследование и эхокардиографию.
(2) Трудности диагностики. У 70-80% больных один из родителей страдает синдромом Марфана. Частота новых мутаций очень высока. Пестрота клинических проявлений и их тяжести создаёт трудности в оценке семейного анамнеза.
При синдроме Марфана или подозрении на него необходимо ежегодно проводить оценку физического развития, офтальмологическое исследование, эхокардиографию. Для предотвращения прогрессирования дилатации устья аорты необходимо назначить b-адреноблокаторы.
- Читать далее "Аутосомно-рецессивные болезни - кистозный фиброз (муковисцидоз)"
Оглавление темы "Врожденные и наследственные болезни у детей":- Причины врожденных пороков (ВП) у детей
- Аутосомно-доминантные болезни - синдром Марфана
- Аутосомно-рецессивные болезни - кистозный фиброз (муковисцидоз)
- Врождённые нарушения обмена веществ - классификация, характеристика
- Нарушения обмена аминокислот и органических кислот: фенилкетонурия, гомоцистинурия, изовалериановая ацидемия
- Нарушения обмена аммониевых соединений - аммиака у детей
- Нарушения метаболизма углеводов: галактоземия, гликогенозы
- Мукополисахаридозы у детей - клиника, диагностика
- Сцепленные с Х-хромосомой болезни: синдром ломкой Х-хромосомы
- Дефицит орнитинкарбамоилтрансферазы у ребенка - клиника, диагностика