
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Нарушения обмена аминокислот и органических кислот: фенилкетонурия, гомоцистинурия, изовалериановая ацидемия
Общая характеристика нарушений обмена аминокислот и органических кислот:
(a) Тип поражения. При заболеваниях этой группы — вследствие недостаточности фермента — происходит блокада метаболизма, в результате чего накапливается субстрат фермента и/или его предшественники. Не менее важно отсутствие продуктов ферментной реакции.
(b) Клиника. Нарушение синтеза необходимых метаболических компонентов обычно ведёт к появлению симптомов уже в раннем детстве. Избыточное накопление предшественников или их метаболитов может нарушать обмен веществ, вызывая, например, тяжёлый ацидоз или алкалоз.
(c) Диагностика. Необходимо определение концентрации аминокислот, органических кислот и их метаболитов в моче или плазме крови.
(d) Терапия. Некоторые из врождённых нарушений обмена веществ не поддаются терапии и приводят к гибели в раннем возрасте или к грубой задержке роста и развития интеллекта. Другие заболевания лечат при помощи диетотерапии или замещения недостающих компонентов. При многих нарушениях обмена органических кислот наблюдают грубые неврологические нарушения, приводящие к летальному исходу в грудном или раннем детском возрасте.
Фенилкетонурия
Фенилкетонурия (ФКУ) — наиболее распространённое и изученное нарушение обмена аминокислот; частота ФКУ составляет 1 на 12.000 живорождённых.
(а) Тип поражения. Дефицит фенилаланингидроксилазы нарушает превращение фенилаланина в тирозин, что приводит к накоплению токсических метаболитов — фенилацетата и фенилацетоуксусной кислоты.
(b) Клиника. В отличие от большинства врождённых нарушений обмена аминокислот, ФКУ не проявляется в грудном возрасте. Без лечения первые симптомы появляются в более старшем возрасте.
(I) Типична умеренная или выраженная задержка интеллектуального развития.
(II) Неврологические нарушения. Мышечный гипертонус и тремор, нарушения поведения.
(III) Гипопигментация развивается в результате блока превращения фенилаланина в тирозин — необходимый промежуточный продукт синтеза меланина,
(c) Профилактика задержки развития интеллекта осуществляется ранней диагностикой и назначением диеты с ограничением потребления продуктов, содержащих фенилаланин.
(I) В США и России должны проводиться обязательные скрининг-тесты, идентифицирующие детей с ФКУ.
(II) Ограничительная диета должна быть введена очень рано (на первом месяце жизни). Для профилактики расстройств интеллекта рекомендуют ограничительную диету в течение всей жизни.
(III) При соблюдении диеты больные ФКУ могут вести нормальный образ жизни. Женщины с ФКУ, не соблюдающие диету, имеют достаточно высокий риск рождения детей с ВП (особенно с микроцефалией), ВПС и нарушениями интеллекта.
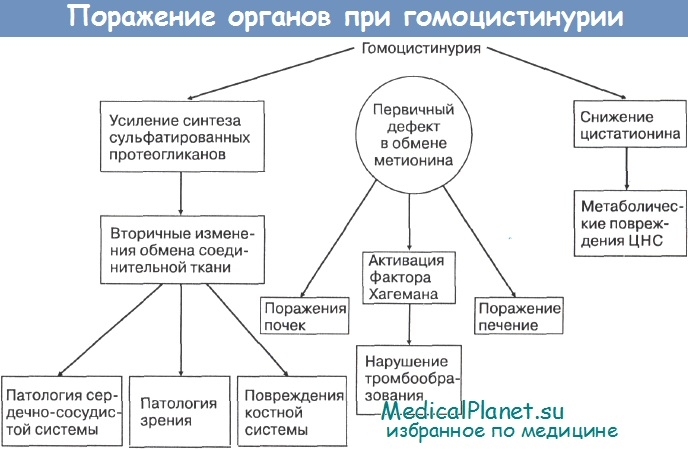
Гомоцистинурия
Несколько различных форм гомоцистинурии. Распространённость по наиболее частой форме — 1 на 100.000 живорождённых детей. Суммарная частота всех форм — значительно больше.
(a) Тип поражения. Дефицит цистатионин-синтазы приводит к накоплению гомоцистина, который экскретируется с мочой. Это генетически гетерогенное заболевание; оно может также развиваться в результате дефицита 5-метилтетрагидрофолата.
(b) Клиника. В грудном возрасте заболевание не проявляется. Основные симптомы детского возраста — аномалии скелета, глаз и пониженный интеллект.
(I) Характерный внешний вид — длинные тонкие конечности и пальцы, сколиоз, деформация грудной клетки, остеопатии — напоминает синдром Марфана.
(II) Часто наблюдается подвывих хрусталика.
(III) У некоторых детей происходит задержка интеллектуального развития.
(IV) Тромбоз сосудов приводит к развитию инсультов и инфарктов в детском возрасте (известно несколько случаев).
(c) Лечение:
(I) При дефиците 5-метилтетрагидрофолата — терапевтические дозы витамина В6.
(II) При В6-резистентной гомоцистинурии — диетотерапия (полное исключение продуктов, содержащих сульфгидрильные группы).
Изовалериановая ацидемия
(а) Тип поражения:
(I) Изовалериановая ацидемия возникает как нарушение обмена предшественников органических кислот при дефиците изовалерил-КоА дегидрогеназы, осуществляющей окислительное декарбоксилирование лейцина.
(II) Из-за выведения с мочой I-карнитина (как поступающего с пищей, так и эндогенного) в форме конъюгата с водорастворимым изовалерил-глицином возникает его вторичная недостаточность.
(b) Клиника:
(I) Дебют заболевания может быть различным: от тяжёлого кетоацидоза новорождённых с энцефалопатией (при отсутствии лечения кетоацидоз прогрессирует и приводит к смерти) до медленно прогрессирующих форм с задержкой развития и эпизодами метаболического ацидоза.
(II) Во время приступов кетоацидоза возможны нейтропения и панцитопения, больные имеют характерный запах (синдром потных ног).
(III) При дефиците i-карнитина возможны миопатии и кардиомиопатия.
(c) Диагностика сводится к определению изовалериановой кислоты, изовалерилглицина и изовалерилкарнитина в моче.
(d) Терапия. Умеренное ограничение белка и дополнительное введение глицина и Z-карнитина. Начало лечения в раннем детском возрасте позволяет предупреждать кетоацидоз и нормализовать развитие.

- Читать далее "Нарушения обмена аммониевых соединений - аммиака у детей"
Оглавление темы "Врожденные и наследственные болезни у детей":- Причины врожденных пороков (ВП) у детей
- Аутосомно-доминантные болезни - синдром Марфана
- Аутосомно-рецессивные болезни - кистозный фиброз (муковисцидоз)
- Врождённые нарушения обмена веществ - классификация, характеристика
- Нарушения обмена аминокислот и органических кислот: фенилкетонурия, гомоцистинурия, изовалериановая ацидемия
- Нарушения обмена аммониевых соединений - аммиака у детей
- Нарушения метаболизма углеводов: галактоземия, гликогенозы
- Мукополисахаридозы у детей - клиника, диагностика
- Сцепленные с Х-хромосомой болезни: синдром ломкой Х-хромосомы
- Дефицит орнитинкарбамоилтрансферазы у ребенка - клиника, диагностика