
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Гликогеноз тип IIa (болезнь Помпе) - причины, диагностика, лечение
Различные виды нейромышечной патологии могут сопровождать многочисленные метаболические заболевания, например лизосомальные или митохондриальные болезни; они представлены в других разделах (см. главы 14, 15 и 20).
Лизосомальные мышечные болезни. Лизосомы представляют собой ограниченные мембраной вакуоли, богатые кислыми гидролазами (производными аппарата Гольджи).
Умеренное поражение скелетных мышц наблюдается при многих состояниях, обусловленных дефицитом лизосомальных ферментов, но зачастую не сопровождается манифестными клиническими симптомами. Умеренная мышечная дисфункция может маскироваться поражением ЦНС или других систем и органов.
При мукополисахаридозах фибробласты эндомизия приобретают типичные включения в вакуолях, но мышечные волокна не изменяются. При фукозидозе, маннозидозе, болезни Фабри в мелких кровеносных сосудах могут обнаруживаться характерные включения аккумулируемого материала в эндотелии и/или клетках гладких мышц, в то время как при ганглиозидозе GM1 поражаются клетки-сателлиты (а не мышечные волокна) и сосудистый эндотелий. Младенческая форма цероидного липофусциноза (болезнь Сантовуори) сопровождается наличием в мышцах характерных включений.
Из лизосомальных болезней, сопровождающихся в неонатальном периоде признаками нейромышечного поражения, мы рассмотрим болезнь Помпе.
Гликогеноз тип IIa (болезнь Помпе) - сцепленное с полом, генетическое заболевание, наследуемое по аутосомно-рецессивному типу (OMIM 232300). Относится к лизосомальным болезням, так как вызывается дефицитом лизосомального фермента (а-1,4-глюкозидазы или кислой мальтазы). Из трех клинических форм болезни (инфантильной, детской и взрослой) в периоде новорожденности встречается инфантильная.
Гликогеноз тип IIa является единственной лизосомальной болезнью накопления с поражением мышц, при которой отмечается выраженная гипотония. При болезни Помпе мышечные волокна прогрессивно увеличиваются в размерах вследствие накопления лизосомального и саркоплазматического гликогена (наряду с аккумуляцией некоторых других аномальных субстанций). Мышечная слабость развивается вследствие комплекса факторов: заполнение пространства отложениями гликогена, миофибриллярные потери, снижение функций мотонейронов в местах накопления гликогена. Патологический процесс начинается до рождения ребенка (внутриутробно).
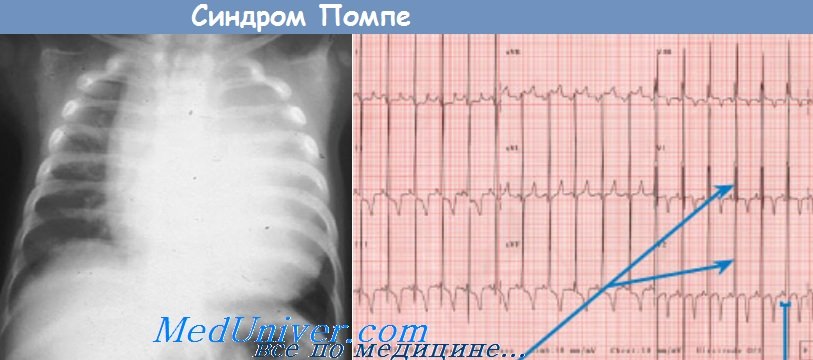
Проявления инфантильной формы болезни Помпе наиболее тяжелы; первыми симптомами являются глубокая мышечная гипотония и кардиомегалия, которые могут появляться в периоде новорожденности. При ЭКГ-исследовании у пациентов обнаруживаются короткие интервалы PR и высокие комплексы QRS во всех отведениях. Нередко к 6-месячному возрасту наступает смерть от сердечной недостаточности.
Наличие гликогеноза тип II следует заподозрить у новорожденных с признаками мышечной гипотонии и кардиомегалии (или сердечной недосточности). В мышечных биоптатах обнаруживаются характерные крупные вакуоли. Диагноз болезни Помпе подтверждается данными исследования концентрации кислой мальтазы в мышцах, лейкоцитах или культурированных фибробластах пациента.
Лечение болезни Помпе не разработано, хотя исследования в этом направлении ведутся. N.J. Mendelsohn и соавт. сообщают об эффективности применения иммуносупрессивной терапии в лечении младенца с инфантильной формой болезни Помпе (заместительный фермент — рекомбинантная человеческая альфа-глюкозидаза, ритуксимаб, метотрексат, человеческий иммуноглобулин для внутривенного введения).
Предполагается, что описываемое лечение позволит постепенно сформировать толерантность к необходимому ферменту и иммуносупрессивная терапия может быть отменена. О перспективах подобной терапии сообщается в недавних публикациях М. Rohrbach и соавт. (2010), Е. Richard и соавт. (2011), С. Angelini и С. Semplicini (2012), RS. Kishani и соавт. (2012), а также Y.H. Messinger и соавт. (2012). Тем не менее прогноз при гликогенозе тип Па пока продолжает оставаться неблагоприятным.
Митохондриальная патология (митохондриопатии) - эта группа метаболических заболеваний представлена в других статьях.
Митохондриальные миопатии проявляются не только мышечной слабостью, но и эпизодически возникающими приступами рвоты, жажды, а также профузного потоотделения. Иногда у таких пациентов наличествует вялая тетраплегия.
Морфологически в мышечных волокнах у новорожденных с митохондриальными миопатия-ми отмечается увеличение липидных включений, реже — некроз, разрастание жировой и соединительной ткани. Метод электронной микроскопии позволяет выявить увеличение размера и/ или количества митохондрий, нарушающих нормальную фибриллярную структуру мышечных клеток.
Разновидности миопатии определяются в зависимости от состояния митохондрий: это мега-кониальная, плеокониальная и гиперметаболическая миопатия.
- Читать "Инфантильные периферические нейропатии - причины, диагностика, лечение"
Оглавление темы "Неврология новорожденных детей":- Синдром ригидного младенца (stiff baby syndrome) - причины, диагностика, лечение
- Синдром Шварца-Джампеля - причины, диагностика, лечение
- Врожденная (семейная) дисавтономия (синдром Райли-Дэя) - причины, диагностика, лечение
- Гликогеноз тип IIa (болезнь Помпе) - причины, диагностика, лечение
- Инфантильные периферические нейропатии - причины, диагностика, лечение
- Фенилкетонурия (ФКУ) у новорожденных - причины, диагностика, лечение
- Болезнь мочи с запахом кленового сиропа (БМЗКС, лейциноз) у новорожденных - причины, диагностика, лечение
- Галактоземия у новорожденных - причины, диагностика, лечение
- Гистидинемия у новорожденных - причины, диагностика, лечение
- Тирозинемия у новорожденных - причины, диагностика, лечение