
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Митохондриальные атаксии - клиника, диагностика
Митохондриальные атаксии представляют собой группу наследственных атактических синдромов, относящихся к обширной группе митохондриальных болезней (митохондриальных энцефаломиопатий).
Митохондриальные болезни — заболевания, обусловленные генетическими и структурно-биохимическими дефектами митохондрий и сопровождающиеся нарушением тканевого дыхания (Вельтищев Ю.Е., Темин П.А., Краснопольская К.Д., Захарова Е.Ю., Иллариошкин С.Н. и др., DiMauro S.).
При митохондриальных болезнях в связи с возникающим системным дефектом энергетического метаболизма поражаются в различной комбинации наиболее энергозависимыс ткани и органы-мишени (мозг, скелетные мышцы и миокард, поджелудочная железа, орган зрения, почки, печень).
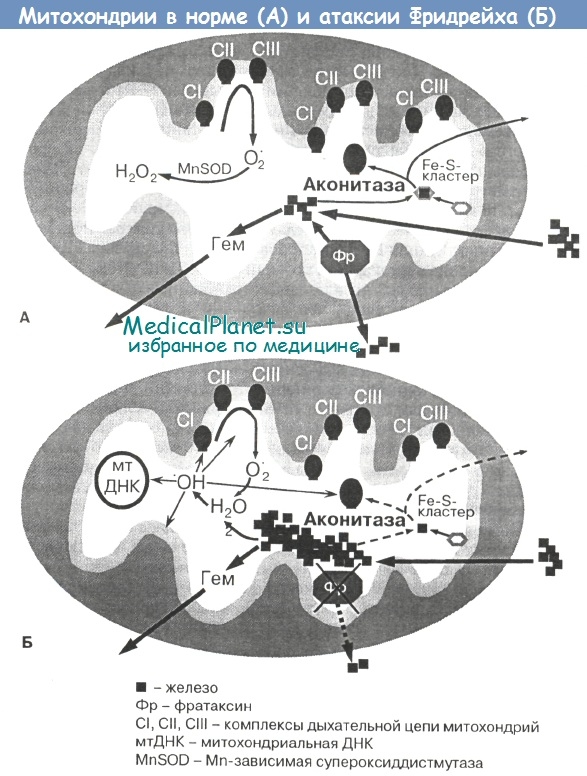
Особенностью функционирования митохондрий является наличие собственного митохондриального генома — кольцевой молекулы ДНК, расположенной внутри данной органеллы и состоящей из 16 569 нуклеотидов. Митохондриальная ДНК (мтДНК) содержит 37 генов, кодирующих синтез 2 видов рибосомальной РНК, 22 видов транспортной РНК (необходимых для синтеза белка в митохондриях), а также 13 белков, входящих в состав I, III, IV и V комплексов дыхательной цепи митохондрий.
Значительная часть белков указанных комплексов, а также все белки II комплекса дыхательной цепи и большое число других митохондриальных белков кодируются генами ядерной ДНК. Таким образом, в обеспечении многообразных биохимических функций митохондрий участвуют белки, кодируемые как ядерными, так и митохондриальными генами.
В соответствии с вышеуказанными особенностями двойного генома митохондрий тип наследования митохондриальных болезней может быть различным. Поскольку вся мтДНК в организме имеет материнское происхождение (из цитоплазмы яйцеклетки), в случае передачи митохондриальной мутации потомству в родословной имеет место материнский тип наследования — когда заболевание проявляется у всех детей больной матери.
Если мутация происходит в ядерном гене, кодирующем синтез митохондриального белка, заболевание передается по классическим мснделсвским законам (чаще всего по аутосомно-доминантному и аутосомно-рецессивному типам). Наконец, при ряде форм митохондриальных болезней мутация мтДНК (обычно делеция) возникает de novo на ранней стадии онтогенеза, приводя к преимущественному накоплению мутантного вида мтДНК в тканях-мишенях; в такой ситуации имеет место спорадический случай болезни.
В результате клеточных делений (митоз, мейоз) молекулы мтДНК в составе митохондрий в случайном порядке переходят в цитоплазму дочерних клеток. В обычной ситуации во всех клетках и тканях организма имеется один и тот же нормальный вид мтДНК -состояние, обозначаемое как гомоплазмия.
При возникновении мутации в мтДНК и дальнейшей амплификации мутантного клона возникает гетероплазмия, т.е. состояние, при котором в клетке (ткани) существует совокупность двух различных популяций мтДНК — нормальной и мутантной. Процентное содержание нормальной и мутантной мтДНК в конкретной ткани может варьировать в широких пределах (от 0 до 100%), что в значительной степени определяет тяжесть соответствующих клинических проявлений болезни.
Для митохондриальных болезней характерен ряд универсальных диагностических тестов, позволяющих подтвердить факт митохондриальной дисфункции и нарушение энергетического метаболизма. К ним относятся:
• лактат-ацидоз — повышение уровня лактата и пирувата в крови и/или цереброспинальной жидкости;
• феномен «рваных красных волокон» — выявление в мышечных биоптатах при специальном окрашивании миофибрилл со своеобразно измененными краями вследствие пролиферации митохондрий и формирования митохондриальных агломератов по периферии мышечного волокна;
• выявляемый при гистохимическом исследовании дефицит цитохром-С-оксидазы в мышечных волокнах;
• электронно-микроскопические признаки патологии митохондрий (аномалии формы и размеров, нарушение конфигурации крист, наличие паркристаллических включений и др.).
Эти признаки, наряду с выявлением специфических звеньев митохондриальной дисфункции и мутаций в митохондриальной/ядерной ДНК, являются важной составной частью диагностического алгоритма при митохондриальных болезнях, в том числе при их атактических фенотипах.
- Читать "Синдром MERRF (Myoclonus Epilepsy with Ragged-Red Fibers) - клиника, диагностика"
Оглавление темы "Атаксии - клиника, диагностика":- Эпизодическая атаксия с пароксизмальным хореоатетозом и спастичностью - клиника, диагностика, лечение
- Аутосомно-доминантная спастическая атаксия (синдром SAX1) - клиника, диагностика
- Аутосомно-доминантная спастическая атаксия с умственной отсталостью (синдром SPAR) - клиника, диагностика
- Заднестолбовая атаксия - клиника, диагностика
- Синдром Герстманна-Штреуслера—Шейнкера - клиника, диагностика
- Наследственные метаболические атаксии - определение, классификация
- Митохондриальные атаксии - клиника, диагностика
- Синдром MERRF (Myoclonus Epilepsy with Ragged-Red Fibers) - клиника, диагностика
- Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) - клиника, диагностика
- Синдром NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) - клиника, диагностика