
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
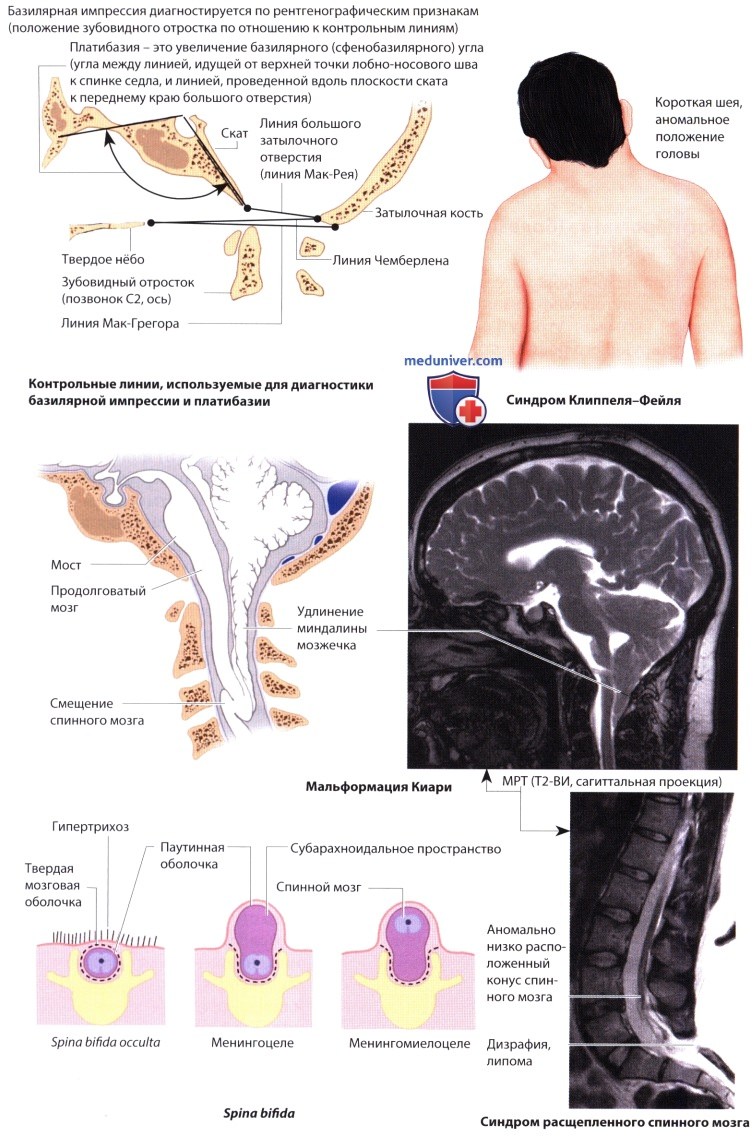
Аномалии развития ЦНС - характеристика кратко
Примеры наследственных и врожденных аномалий развития ЦНС представлены в таблице ниже.


а) Врожденные двигательные аномалии. Двигательные нарушения, которые начинаются в раннем возрасте и называются детским церебральным параличом или церебральным параличом, обусловлены повреждением головного мозга до, во время или после родов. Они проявляются непрогрессирующими, но не обязательно однотипными двигательными и постуральными нарушениями. Лежащее в их основе поражение мозга обычно имеет многофакторное происхождение. К пренатальным причинам относятся хромосомные нарушения, инфекции, гипоксия, несовместимость крови матери и плода по группе крови (или резус-фактору).
К перинатальным причинам относятся гипоксия, мозговое кровоизлияние, побочные эффекты лекарств и желтуха новорожденных. К постнатальным - менингоэнцефалит, инсульт, опухоль мозга, метаболические нарушения и травма. Менее 4 баллов по шкале Апгар в течение более 1 мин сочетаются с повышенным риском двигательных нарушений.
Недостаточность спонтанных движений, аномальный характер движений становятся заметными сразу после рождения, а задержка вставания на ноги и первых шагов - по мере развития ребенка. Среди аномалий часто встречаются центральный парез (геми-, пара-, тетрапарез), спастич-ность, атаксия и хореоатетоз. Кроме этого, могут наблюдаться задержка умственного развития, эпилептические судороги, поведенческие нарушения (беспокойство, импульсивность, отсутствие концентрации), нарушения зрения, слуха и речи. Двигательные нарушения приводят к деформациям костей и суставов («конская стопа», контрактуры, сколиоз, вывих тазобедренного сустава). Более подробно о диагностике см. отдельную статью на сайте - просьба пользоваться формой поиска выше.
Необходимо как можно раньше начинать занятия лечебной физкультурой, трудовую и речевую терапию, а также тренировку перцепции. Для лечения спастичности некоторых мышц (динамическая «конская стопа», приводящих мышц бедра, сгибателей предплечья) применяют ботулинический токсин. Прочие меры включают хирургические манипуляции (коррекция деформации суставов), коррекцию зрения, помощь в общении, занятия спортом, консультации диетолога.
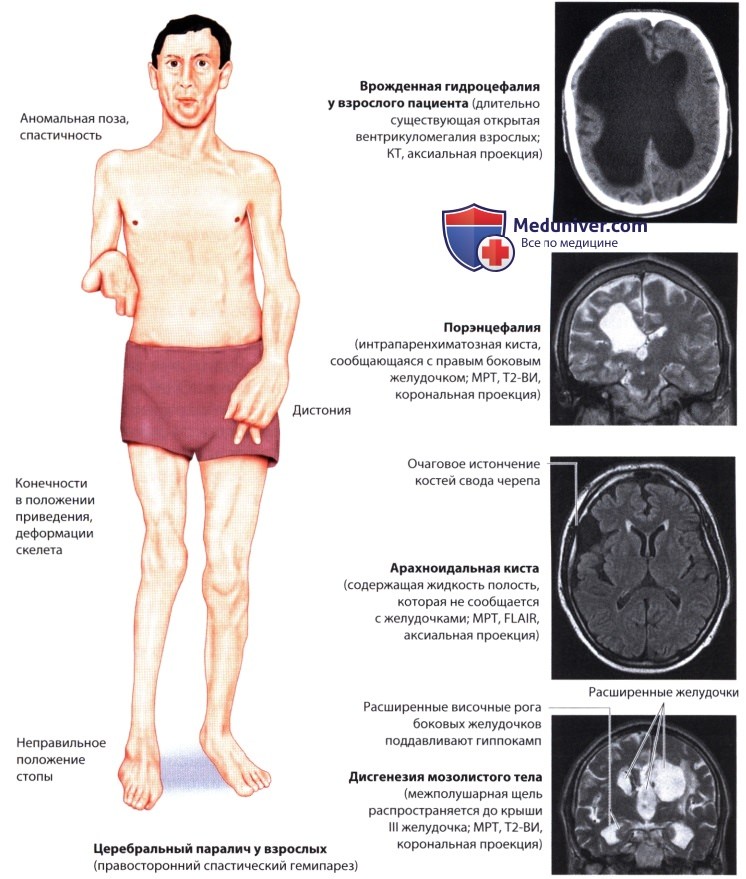
б) Гидроцефалия. Причинами гидроцефалии могут быть стеноз сильвиева водопровода, мальформации Денди-Уокера и Киари, инфекции (токсоплазмоз, вентрикулит), кровоизлияние и обтурирующие опухоли (коллоидная киста III желудочка, опухоль средней линии).
Если черепные швы еще не срослись, врожденная обструктивная гидроцефалия приводит к увеличению размеров головы (макроцефалия) с лобными выступами, формирующимися в результате постоянно повышенного ВЧД. Дополнительными признаками являются выступающие вены скальпа, истончение скальпа и парез взора вверх (при этом глаза все время направлены вниз, а между радужкой и верхним веком видна склера).
Необходимо регулярно измерять окружность головы, так как это более полезный индикатор врожденной гидроцефалии, чем клинические признаки повышенного ВЧД. Последние часто не слишком выражены у маленьких детей и могут маскироваться беспокойством, недостаточной прибавкой массы тела, криком и задержкой психомоторного развития. Острая гидроцефалия обычно требует нейрохирургического вмешательства.
в) Порэнцефалия. Повреждение головного мозга в позднем внутриутробном или раннем постнатальном периоде может привести к образованию кист или полостей (от греч. poros - отверстие, проход). При порэнцефалии полости сообщаются с желудочками или субарахноидальным пространством. Причиной часто служит инфаркт, кровоизлияние, травма или инфекция. Порэнцефалические кисты редко сопровождаются повышением ВЧД. Крупные кисты отражают значительную утрату ткани мозга. Крайние случаи называются гидранэнцефалией (полное или почти полное отсутствие больших полушарий). Клинические признаки зависят от размера, локализации и причины порэнцефалии. Это могут быть двигательные нарушения, эпилептические судороги и ментальные нарушения.
г) Арахноидальные кисты. Эти аномалии развития мозговых оболочек формируются при расщеплении паутинной оболочки. Они возникают в различных местах. Некоторые кисты сообщаются с субарахноидальным пространством. Многие из них, даже крупные, не проявляются клинически. В редких случаях они могут препятствовать току ЦСЖ (арахноидальные кисты средней линии или инфратенториальные арахноидальные кисты), а также вызывать новые симптомы вследствие кровоизлияния внутрь кисты, ее роста или разрыва. Арахноидальные кисты лечат оперативным путем с помощью дренирования, эксцизии, фенестрации, эндоскопической фенестрации или шунтирования.
д) Агенезия мозолистого тела. Гипоплазия или агенезия мозолистого тела встречается как изолированная аномалия, так и в сочетании с другими нарушениями развития (мальформация Киари, гетеротопия, хромосомные нарушения, синдром Айкарди → детские спазмы, микрофтальмия, хориоретинопатия, аномалии ребер и позвоночника). Изолированную агенезию мозолистого тела иногда обнаруживают случайно при КТ или МРТ.
Кистозные деформации прозрачной перегородки (cavum septi pellucidi - полость прозрачной перегородки, cavum vergae - полость Верге) иногда могут препятствовать току ЦСЖ и вызывать повышение ВЧД.
Факоматозы - это группа редких наследственных заболеваний, характеризующихся поражением нервной системы, глаз, кожи, а также предрасположенностью к развитию опухолеподобных мальформаций (гамартом) и злокачественных опухолей (гамартобластом). Факоматозы выявляются в детском и подростковом возрасте.
е) Нейрофиброматоз (болезнь фон Реклингхаузена). Мутация гена нейрофиброматоза 1-го типа (NF1, 17q11.2, аутосомно-доминантное наследование) вызывает утрату нейрофибромина (регулирующего рост клеток), а мутация гена нейрофиброматоза 2-го типа (NF2, 22q 12.2, аутосомно-доминантное наследование) - утрату нейрофибромина 2 (мерлин, или шванномин; контролирует различные функции клеток).
Клинические проявления. Характерные очаги нейрофиброматоза 1-го типа обнаруживаются в коже (ранняя стадия: пятна цвета кофе с молоком, пигментация в подмышечной/паховой области; поздняя стадия: нейрофибромы/плексиформные нейрофибромы), в глазах (узелки Лиша - меланоцитарные гамартомы радужки, глиома зрительного нерва) и в костях (кисты, патологические переломы, дефекты костей черепа, сколиоз). Дополнительными нарушениями могут быть сирингомиелия, гидроцефалия, эпилептические припадки, преждевременное половое созревание или феохромоцитома. Отличительным признаком нейрофиброматоза 2-го типа является двусторонняя невринома слухового нерва, которая сопровождается прогрессирующей глухотой.
Кожные проявления бывают редко; чаще встречаются другие опухоли нервной системы (нейрофиброма, менингиома, шваннома, глиома). Типичный признак нейрофиброматоза 2-го типа у детей - субкапсулярная катаракта.
Лечение. Хирургическое удаление опухолей. Комбинированное лечение злокачественных опухолей (операция, лучевая терапия, химиотерапия).
ж) Комплекс туберозного склероза (синдром Бурневилля-Прингла). Фенотипы при мутациях гена TSC1 (9q34.13, белок гамартин, наследование аутосомно-доминантное) и гена TSC2 (16р13.3, белок туберин, наследование аутосомно-доминантное) одинаковы. Оба белка работают как супрессоры опухолей.
Клинические проявления. Эпилептические припадки (инфантильные спазмы и гипсаритмия - синдром Веста, позднее фокальные и генерализованные припадки) сочетаются с поражением кожи (ранние проявления: гипопигментированные линейные пятна, видимые при освещении ультрафиолетовым светом; поздние проявления: аденомы сальных желез, подногтевые ангиофибромы, утолщение и уплотнение кожи в области поясницы), глазными заболеваниями (гамартома сетчатки) и опухолями (сердечная рабдомиома, почечная ангиомиолипома, кисты). Могут отмечаться заметная задержка умственного развития и поведенческие нарушения (вокальная и моторная стереотипия, психомоторное беспокойство). КТ и МРТ выявляют перивентрикулярную кальцификацию, поражение коры и опухоли.
Лечение. Симптоматическое (противоэнилептическая терапия).
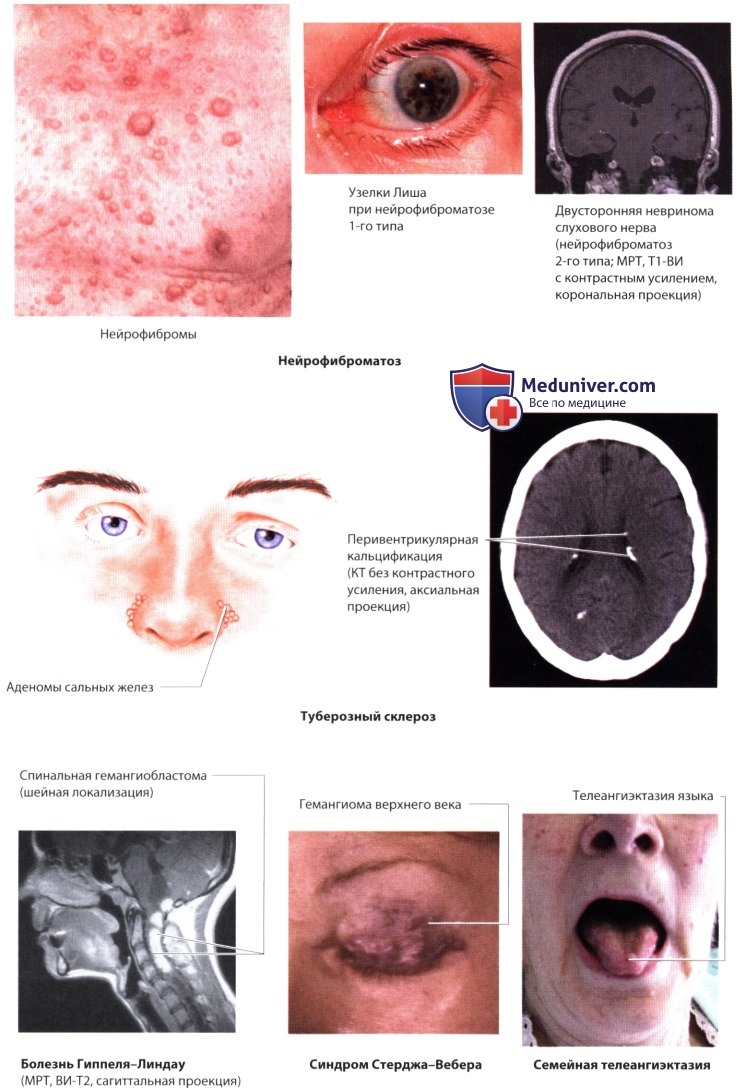
з) Болезнь Гиппеля-Линдау. Мутация гена VHL (ген-супрессор опухолей, 3р25.3, наследование аутосомно-доминантное).
Клинические проявления. Кистозная гемангиобластома мозжечка вызывает головную боль, вертиго и атаксию, иногда гидроцефалию вследствие компрессии IV желудочка. Гемангиомы могут развиваться и в спинном мозге. Часто страдают глаза (ангиоматоз сетчатки → отслойка сетчатки), почки (кисты, карцинома), надпочечники (феохромоцитома), поджелудочная железа (множественные кисты) и придатки яичек (цистаденома).
Лечение. Регулярный скрининг органов и систем, потенциально подверженных патологическим изменениям. Это способствует раннему выявлению и удалению опухолей и предупреждению сосудистых осложнений.
и) Кожный ангиоматоз с поражением ЦНС:
1. Синдром Стерджа-Вебера (энцефало-тригеминальный ангиоматоз). Одно- или двусторонние «винные пятна» (port wine stains) - другое название «пламенеющие невусы» (naevus flammeus) - обнаруживаются при рождении и могут быть как локализованными (типично на верхних веках и лбу; в этом случае вероятно вовлечение в процесс головного мозга), так и распространенными (вся голова или тело). Не все кожные гемангиомы сопровожаются поражением мозга.
2. Наследственная геморрагическая телеангиэктазия (синдром Ослера-Вебера-Рандю). Мутация гена ННТ (белок эндоглин, 9q34.11, наследование аутосомно-доминантное). Телеангиэктазии (сосудистые аномалии) кожи, слизистых оболочек, ЖКТ, мочеполового тракта и ЦНС вызывают повторные кровотечения (носовое, желудочное, легочное, гематурия, мозговое кровоизлияние, анемия). Артериовенозный шунт в легких может быть причиной цианоза и полицитемии.
- Читать "Сбор анамнеза у неврологического пациента"
Редактор: Искандер Милевски. Дата публикации: 9.4.2020