
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Трудности дифференциации наследственных и врожденных заболеваний у детей
Значительные дифференциально-диагностические трудности возникают и при обследовании детей, у которых подозреваются моногенные болезни - наследственные нарушения обмена веществ (аминокислот, углеводов, липидов, соединительной ткани, витаминов, минералов и др.). При этом врач постоянно сталкивается с проблемой фенотипического сходства, при котором разные по своему генезу заболевания клинически весьма похожи. Так дети с витамин D-peзистентным рахитом или синдромом де Тони - Дебре- Фанкони напоминают больных с витамин D-дефицитным рахитом.
А легочная форма муковисцидоза у детей чаще всего маскируется под хроническую пневмонию. Это обусловлено тем, что разные по своему происхождению наследственные заболевания часто имеют общие симптомы: умственную отсталость, костные аномалии, нарушения питания, патологию зрения и слуха, нарушения кишечного всасывания и др. Действительно, умственная недостаточность - ведущий симптом при 60 наследственных нарушениях обмена веществ, при большинстве хромосомных заболеваний и врожденных синдромов. Костные аномалии доминируют в клинической картине более 200 заболеваний наследственной природы (рахитоподобные заболевания, несовершенный остеогенез, синдром Марфана, мукополисахаридозы, артрогрипоз и др.).
Патология зрения входит в симптомокомплекс 280 врожденных и наследственных заболеваний (ганглиозидозы, галактоземия, синдром Марфана, гомоцистинурия, синдром Лоуренса - Муна - Барде - Бидля и др.). Нарушения физического развития обнаруживаются при большинстве врожденной и наследственной патологии и проявляются как в задержке, так и в ускоренном темпе роста (синдром Марфана, прогерия, нанизм и др.).
При большинстве наследственных заболеваний в патологический процесс вовлекаются многие органы и системы. Например, при синдроме Марфана обнаруживаемый симптомокомплекс обусловлен нарушениями обмена соединительной ткани (изменения в опорно-двигательном аппарате, сердечно-сосудистые расстройства, дисгармоничность физического развития, патология зрения и ЦНС).
При рахитоподобных заболеваниях в патологический процесс вовлекаются почки, скелет, нервно-мышечная система, кишечник, железы внутренней секреции и др. Особенно тяжелый по своим последствиям симптомокомплекс обнаруживается при синдроме Лоуренса - Муна - Барде - Бидля: эндокринные расстройства (ожирение, гипогенитализм), тяжелая патология зрения, умственная отсталость, пороки развития мочевыделительной системы и др. Следовательно, врач сталкивается с довольно «пестрой» клинической картиной, которая складывается из первичных и вторично возникающих симптомов.
Опыт свидетельствует о том, что так называемое фенотипическое сходство, о котором так часто упоминается в литературе, на самом деле чаще всего лишь кажущееся. Это происходит потому, что из целого симптомокомплекса нередко искусственно вычленяют отдельные симптомы и придают им особое значение. Действительно, в клинической картине наследственных заболеваний обмена веществ среди многих обнаруживаемых симптомов нередко доминирует один какой-либо серьезный дефект (умственная отсталость, слепота, глухота, скелетные аномалии и др.), вызывающий особые трудности при адаптации больного к окружающей среде. Этот дефект настолько очевиден для родителей больного ребенка и для врача, что многие другие нарушения как бы исчезают из поля зрения и приводят к неправильной интерпретации врачом наблюдаемых у больного расстройств. Так, умственная отсталость - основной симптом при многих врожденных и наследственных заболеваниях -является поводом для диагностики олигофрении и направлении ребенка к психиатру.
Дети с дефектами зрения, входящими в симптомокомплекс наследственного заболевания, обычно наблюдаются и лечатся у офтальмологов. Дети с рахитоподобными заболеваниями особенно часто состоят на учете и лечатся у ортопедов по поводу костных деформаций или у педиатров, как больные с последствиями витамин D-дефицитного рахита и т.п. Таким образом, указанные тяжелые инвалидизирующие расстройства, доминируя в клинической картине, как бы нивелируют все остальные признаки болезни и тем самым приводят к недооценке состояния больного в целом и неправильной диагностике.
В то же время очевидно, что помимо общих, объединяющих разные генетически гетерогенные заболевания признаков, часто имеются и специфические, делающие каждый симптомокомплекс своеобразным. Следовательно, в дифференциальной диагностике важная роль должна отводиться не отдельным симптомам (как бы тяжелы они ни были), а ихсочетанию. В качестве примера, иллюстрирующего это принципиально важное положение, можно привести такие заболевания, как фенилкетонурия (ФКУ), гомоцистинурия, галактоземия и гистидинемия.
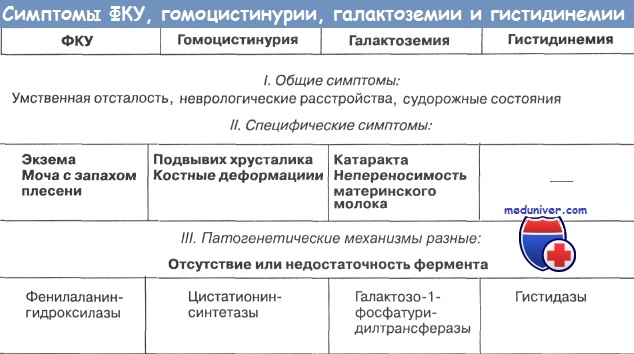
Эти генетически гетерогенные заболевания фенотипически сходны только по наличию общих симптомов (поражение ЦНС). Если же принимать во внимание симптомокомплекс каждого заболевания в целом, то фенотипического сходства не будет. Так, при гомоцистинурии поражение ЦНС сочетается с подвывихом хрусталиков и костными деформациями, при галактоземии - с катарактой, гепатоспленомегалией и диспептическими нарушениями.
Однако на этом примере становятся особенно демонстративными трудности диагностики и ведущая роль биохимических исследований при аминоацидопатиях: снижение или полное отсутствие гистидазы в коже при гистидинемии, обнаружение при ФКУ высоких концентраций фенилаланина в крови, наличие в моче фенилпировиноградной кислоты и снижение или отсутствие фенилаланингидроксилазы в печени; при гомоцистинурии - повышение концентрации метионина в крови, появление в моче гомоцистина и смешанных дисульфидов гомоцистеина - цистеина, снижение или отсутствие активности фермента цистатионинсинтетазы и т.д.
Однако это положение может быть справедливым только при наличии полного симптомокомплекса болезни. В повседневной практике врач встречается с такой ситуацией, когда у больного обнаруживается только часть характерных для заболевания симптомов. Полнота симптомокомплекса зависит от различных причин: неполной пенетрантности гена, экспрессивности признака, степени тяжести ферментативной недостаточности, характера течения патологического процесса.
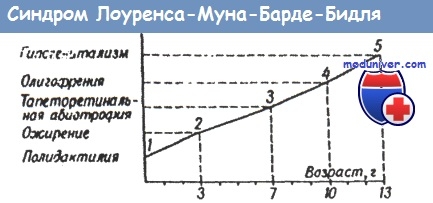
При прогредиентном течении патологического процесса большое значение приобретает возраст больного. В раннем возрасте у ребенка обычно имеется неполный симптомокомплекс и это затрудняет процесс дифференциальной диагностики, значительно расширяет количество нозологических форм, которые следует принять во внимание. Так, синдром Лоуренса - Муна - Барде - Бидля полностью формируется у детей к 12-13 годам. Предполагаемый диагноз на первом году жизни - полидактилия, синдром Карпентера, синдром Эллиса - ван - Кревельда, синдром Мекеля - Грубера, синдром Робинсона.
К концу второго года жизни полидактилия может сочетаться с избыточной массой тела, которой обычно не придают особого значения. Лишь при появлении изменений со стороны сетчатой оболочки глаз возникают подозрения на наличие тяжелых страданий (нейролипидозы, адипозогенитальное ожирение, тапеторетинальная абиотрофия, синдром Коэна, синдром Коккейна, синдром Прадера-Вилли). Обнаружение в последующем умственной отсталости и гипогенитализма резко сужает рамки дифференциального диагноза, так как формирование синдрома полностью заканчивается.
Аналогичная ситуация происходит и при формировании синдрома Марфана: арахнодактилия обнаруживается при рождении ребенка (предполагаемый диагноз: гомоцистинурия, синдром Стиклера, синдром Горлина), глазная патология - на третьем году жизни (предполагаемый диагноз: гомоцистинурия, синдром Вейла - Маркезани, синдром Элерса - Данлоса), деформация грудной клетки и сердечно-сосудистые расстройства - к семи-восьми годам, а к девяти годам формирование синдрома заканчивается.
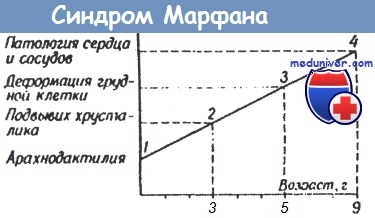
Сроки манифестации и формирования симптомокомплекса в значительной мере зависят от характера первичного генетического дефекта. При неполном метаболическом блоке наблюдается более медленное течение заболевания и менее тяжелые поражения ЦНС, чем при полном. Так, у детей с ФКУ и гистидинемией при полном метаболическом блоке и отсутствии фенилаланингидроксилазы или гистидазы происходит очень быстрое формирование тяжелого церебрального дефекта, а при неполном метаболическом блоке не только удлиняются сроки манифестации психоневрологических расстройств, но и уменьшается степень их тяжести.
Из этих примеров видно, как велика роль тщательного клинического обследования больного с наследственной патологией. Прежде всего это касается получения объективной информации о состоянии тех органов и систем, которые особенно часто вовлекаются в патологический процесс: ЦНС, зрение, слух, кожа, мочеполовая, сердечно-сосудистая и бронхолегочная системы, скелет и др. Особое диагностическое значение следует придавать сочетанию наблюдаемых симптомов. При этом обращается внимание на сроки манифестации отдельных болезненных симптомов, так как часто они появляются в определенной последовательности.
В процессе дифференциальной диагностики можно выделить два основных этапа. Первый этап - это обследование больного на долабораторном уровне (сбор генеалогических сведений, жалоб, врачебный осмотр и др.). Основной задачей при этом является установление предварительного диагноза и перечня тех заболеваний, с которыми следует проводить дифференциальный диагноз. На втором этапе в обследовании больного целенаправленно используются те или иные методы лабораторной диагностики (микробиологические, биохимические, иммунологические, цитогенетические и др.).
- Рекомендуем далее ознакомиться со статьей "Сбор анамнеза при наследственных и врожденных заболеваний у детей"
Оглавление темы "Признаки наследственных и врожденных заболеваний":- Наследственные и врожденные заболевания в период новорожденности
- Наследственные и врожденные заболевания в период новорожденности
- Наследственные, врожденные заболевания у детей раннего и подросткового возраста
- Трудности дифференциации наследственных и врожденных заболеваний у детей
- Сбор анамнеза при наследственных и врожденных заболеваний у детей
- Типы наследования наследственных заболеваний у детей
- Факторы риска наследственных и врожденных заболеваний при беременности
- Нарушения роста, развития плода при наследственных и врожденных заболеваниях
- Рост, развитие детей при наследственных и врожденных заболеваниях
- Болезни нервной системы детей при наследственных и врожденных заболеваниях