
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдромы серебристых волос: синдром Чедиака-Хигаси
Синдромы «серебристых волос» включают группу аутосомно-рецессивных заболеваний, при которых наблюдается необычный специфический металлический отблеск на волосах и часто на коже.
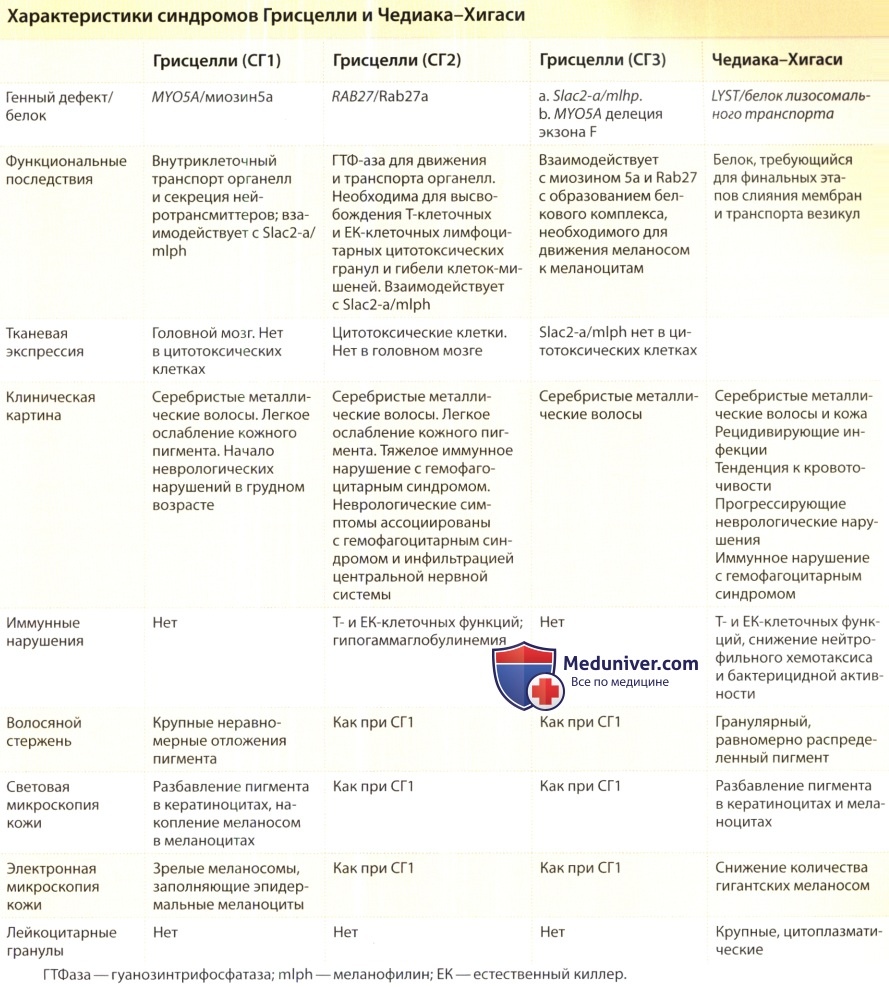
Из всех этих заболеваний только при синдромах Чедиака-Хигаси и Грисцелли отмечается сопутствующий иммунодефицит. Другой синдром с «серебристыми» волосами — синдром Элейалда — не сопровождается иммунодефицитом.
Синдром Элейалда может быть аллельным с миозин 5а-дефицитной формой синдрома Грисцелли (СГ) с тяжелой сочетанной дисфункцией ЦНС.
Синдром Чедиака-Хигаси - краткий обзор:
- Аутосомно-рецессивное заболевание транспорта везикул, приводящее к образованию гигантских органелл, в том числе меланосом, лейкоцитарных гранул и плотных гранул тромбоцитов.
- Мутации в гене LYST.
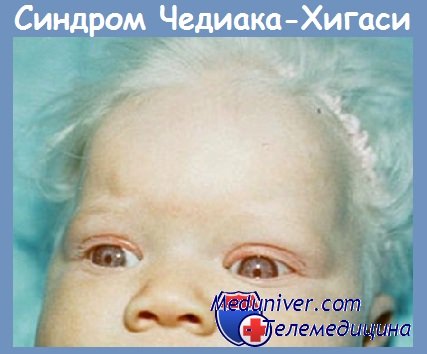
- Серебристые волосы, часто гиперпигментация на носу и ушах, различная степень фотофобии и нистагма.
- Рецидивирующие гнойные инфекции и легкий кровоточивый диатез.
- «Фаза акселерации» с панцитопенией и органомегалией вследствие лимфогистиоцитарной инфильтрации (без трансплантации гематопоэтических стволовых клеток — фатальное течение).
- Прогрессирующая неврологическая дегенерация.
- Лечение: трансплантация.

А. Серебристый отблеск волос у чернокожего грудного ребенка; у пациента отмечается темная пигментация глаз и кожи.
Обратите внимание на усиление пигментации на завитке ушной раковины.
Б. Мелкие, равномерно расположенные пигментные гранулы в волосах при синдроме Чедиака-Хигаси.
В. Более крупные скопления неравномерно распределенной пигментации в волосах пациента с синдромом Грисцелли.
а) Эпидемиология. Синдром Чедиака-Хигаси (СЧХ) представляет собой редкое аутосомно-рецессивное заболевание. Среди родителей больных детей нередки близкородственные браки. В мире зарегистрировано около 300 случаев.
б) Этиология и патогенез. Причиной синдрома Чедиака-Хигакси являются мутации в гене LYST, локализующегося на хромосоме 1q42 и кодирующего белок, необходимый для последних этапов транспортировки везикул и секреции. Ген LYST экспрессируется в лизосомах и других секреторных органеллах (меланосомы, цитолитические гранулы и плотные гранулы тромбоцитов). Характерные гигантские гранулы, как полагают, являются результатом изменения созревания и слияния гранул.
Увеличенные меланосомы не могут транспортироваться к кератиноцитам. ЕК-клетки и цитотоксические Т-лимфоциты не высвобождают протеолитические ферменты, необходимые для уничтожения клеток-мишеней. Цитотоксический Т-лимфоцитарный антиген попадает в «ловушку» в патологически увеличенных везикулах, а не на поверхности клеток, таким образом, может нарушаться регуляция Т-клеточной активации, что повышает риск лимфопролиферативного заболевания. Секреция плотных гранул тромбоцитов из запасных пулов, что необходимо для нормальной коагуляции, задерживается. Часто отмечается сочетанное снижение хемотаксиса нейтрофилов и моноцитов, а также задержка внутриклеточного уничтожения микроорганизмов.
в) Клиническая картина. Первые симптомы обычно проявляются в грудном или раннем детском возрасте. Пигментные нарушения встречаются у 75% пациентов, в частности — серебристый оттенок волос. Гипопигментация глаз может приводить к фотофобии, часто встречаются страбизм и нистагмы. Острота зрения, как правило, не снижается. Кожа обычно светлая, но могут отмечаться участки синевато-серой (аспидной) пигментации. Как правило, кожные проявления отсутствуют, однако могут наблюдаться темные серовато-серые участки пигментации и диффузные пятнистые участки гипопигментации на загорелой коже или у темнокожих пациентов.
Инфекции могут персистировать на протяжении всей жизни пациента, начиная с грудного возраста. Чаще всего инфекции поражают кожу, легкие и дыхательные пути с преобладанием таких возбудителей, как S. aureus, S. Pyogenes и pneumococcus. Были описаны случаи глубоких изъязвлений, напоминающих гангренозную пиодермию.
К неврологическим симптомам относятся слабость, нейропатия черепных и периферических нервов и прогрессирующая неврологическая деградация. У пациентов с синдромом Чедиака-Хигаси также наблюдались признаки умеренных нарушений коагуляции, легко образуются синяки, появляется петехиальная сыпь и отмечаются кровотечения из слизистых оболочек, несмотря на нормальное число тромбоцитов.
Высокоинформативным признаком является обнаружение крупных цитоплазматических гранул в лейкоцитах крови. В волосяных стержнях отмечается равномерное распределение пигментных гранул, в отличие от синдрома Грисцелли, при котором выявляются крупные пигментные скопления. В биоптате кожи определяется ослабление пигмента как в меланоцитах, так и в кератиноцитах, при электронной микроскопии выявляются гигантские меланосомы.
г) Прогноз, клиническое течение и лечение синдрома Чедиака-Хигаси (СЧХ). К позднему детскому возрасту развиваются гемофагоцитарный синдром и фаза акселерации заболевания с признаками лимфомы. Гематофагоцитарный синдром характеризуется распространенной инфильтрацией внутренних органов атипичными лимфоидными и гистиоцитарными клетками. Эта лимфомоподобная стадия провоцируется вирусами, в частности, ЭБВ-инфекцией. Типичными симптомами являются гепатоспленомегалия, лимфаденопатия, панцитопения, желтуха, лихорадка, лейкозоподобный гингивит и псевдомембранозное отторжение слизистой оболочки щек.
Гемофагоцитарный синдром, сочетанный с лимфогистиоцитарной пролиферацией при синдромах «серебристых волос», необходимо дифференцировать от массивной лимфопролиферации при других иммунодефицитных состояниях, в частности, Х-сцепленного лимфопролиферативного синдрома (см. раздел «Нарушения гуморального иммунитета»), семейного гемофагоцитарного лимфогистиоцитоза, аутоиммунного лимфопролиферативного синдрома и иммунодефицита вследствие мутаций в IL-2R. Семейный гемофагоцитарный лимфогистиоцитоз представляет собой группу аутосомно-рецессивных заболеваний с гемофагоцитозом и отсутствием цитотоксичности ЕК-клеток без проведения ранней трансплантации стволовых клеток, имеющих тенденцию к фатальному течению.
Данное состояние также может быть результатом мутации в гене, кодирующем перфорин, являющийся наиболее важным внутриклеточным белком для NK-клеток и цитотоксических Т- лимфоцитов, или в UNC13D, кодирующем Munc 13-4, белок, запускающий секрецию цитолитических вакуолей. Мутации в некоторых генах могут привести к аутосомно-рецессивному или доминантному аутоиммунному лимфопролиферативному синдрому, также известному как синдром Кэнейла-Смита. Необходимо рассмотреть следующие гены: ген, кодирующий Fas или CD95 (TNFRSF6), рецептор апоптоза клеточной поверхности, и его лиганд (TNFSF6), и каспазу белков 8 и 10, протеазы в каскаде, приводящем к апоптозу.
Эта группа расстройств характеризуется аутоиммунными нарушениями (цитопенией, лейкоцитокластным васкулитом, красной волчанкой) и повышенным риском развития лимфомы, что связано с увеличением циркулирующих CD4-/CD8- α/β Т клеток. Иммунодефицит с обширным лимфоцитарным инфильтратом висцеральных органов также является проявлением аутосомно-рецессивной мутации в a-цепи рецептора интерлейкина-2. У данных пациентов наблюдаются бактериальные, вирусные и грибковые инфекции из-за раннего апоптоза формирующихся тимусных Т-лимфоцитов.
Средний возраст смерти пациентов с синдромом Чедиака-Хигаси составляет шесть лет, причиной обычно служит крайне тяжелая инфекция или кровотечение во время лимфомоподобной фазы акселерации. Пренатальная диагностика проводилась при исследовании волос из биопсийного материала с волосистой части кожи головы плода и лейкоцитов из образцов фетальной крови.
Методом выбора при лечении пациентов с синдромом Чедиака-Хигаси является выполнение ранней трансплантации, корректирующей иммунологический статус, но не влияющей на пигментные нарушения и не препятствующей развитию неврологических симптомов, которые ухудшаются с возрастом.
Для контроля фазы акселерации применялись ацикловир, высокие дозы внутривенного гаммаглобулина, винкристин, циклоспорин и преднизолон, однако эта стадия заболевания обычно заканчивается фатально.
Аскорбиновая кислота корректирует микротубулярные дефекты in vitro, однако доказанных клинических положительных результатов получено не было. Некоторыми авторами указывалось, что интерферон может частично восстанавливать функцию ЕК-клеток.
- Рекомендуем далее ознакомиться со статьей "Синдромы серебристых волос: синдром Грисцелли"
Редактор: Искандер Милевски. Дата публикации: 26.12.2018