
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Лобно-височная деменция. Диагностика лобно-височной деменции.
Лобно-височная деменция представляет собой особую форму дегенеративных пресенильных деменции, характеризующуюся атрофией лобных и височных отделов больших полушарий мозга. Существует несколько фенотипических вариантов лобно-височной деменции, описанных в литературе под различными названиями: «деменция лобного типа», «лобно-височная деменция с паркинсонизмом», «деменция с афазией и расторможенностью», «комплекс расторможенность-деменция-паркинсонизм-амиотрофии» и т.п.
Одним из вариантов лобно-височной деменции является болезнь Пика, при которой обнаруживаются характерные гистологические маркеры и преимущественное вовлечение в дегенеративный процесс префронтальной коры, передних и медиальных отделов височной коры. Согласно современным представлениям, все указанные синдромы относятся к группе патогенетически близких нейродегенеративных заболеваний, составляющих единый континуум взаимно перекрывающихся клинико-морфологических форм.
Истинная частота лобно-височной деменции окончательно не установлена; по некоторым оценкам, она может составлять 5 случаев на 1 000 000 населения, или около 30 случаев на 100 000 в возрастной популяции старше 60 лет. Предполагается, что лобно-височная деменция составляет от 10 до 20% всех случаев дегенеративных деменции.
В развитии лобно-височной деменции значительную роль играют генетические механизмы: не менее чем в 30—50% случаев заболевания имеется положительный семейный анамнез, свидетельствующий об аутосомно-доминантном типе наследования. Предполагается, что значительная часть спорадических случаев лобно-височной деменции может быть связана с неполной пенетрантностью мутантного гена либо с недостаточностью сведений о семейном анамнезе (вследствие позднего возраста начала заболевания его достоверная регистрация в ряду поколений весьма затруднена).
Наиболее высокая семейная отягощенность характерна для случаев лобно-височной деменции, сочетающихся с паркинсонизмом. В семейных случаях лобно-височной деменции с паркинсонизмом патологический ген картирован на длинном плече 17-й хромосомы. Совсем недавно, в 1998—99 годах, было показано, что данный ген кодирует синтез /яду-протеина, играющего важную роль в функционировании микротубулярного аппарата нейронов.
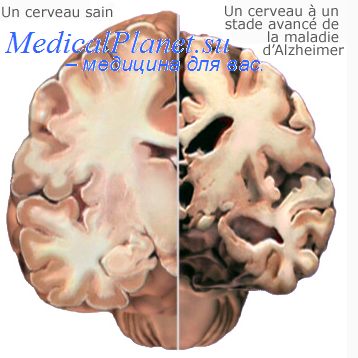
Морфологическая картина лобно-височной деменции характеризуется, главным образом, атрофией коры больших полушарий с гибелью нейронов, поверхностным спонгиозом и распространенным глиозом. Атрофия затрагивает преимущественно лобные и (в меньшей степени) височные и теменные доли полушарий мозга. Аналогичные дегенеративные изменения с большим постоянством обнаруживаются также в базальных ганглиях, черной субстанции, зубчатом ядре и коре мозжечка, гиппокампе, передних рогах спинного мозга.
Характерные для болезни Пика баллонообразные клетки с центральным хроматолизом (клетки Пика) и аргирофильные включения (тельца Пика), обнаруживаются не более чем в 20% случаев лобно-височной деменции.
Заболевание начинается на 5—7 десятилетии жизни и проявляется прогрессирующей деменцией лобного типа, сопровождающейся двигательными нарушениями. Типичными ранними симптомами являются апатия, речевая аспонтанность, эмоциональная тупость, персеверации. Весьма характерны расторможенность и утрата нравственных установок, склонность к асоциальным поступкам. На более поздней стадии болезни нарастает оскудение моторики и мышления, снижается уровень суждений, способность оперировать абстрактными понятиями и конструировать адекватную программу действий.
Наблюдаются поведенческие стереотипии, моторная афазия, аграфия, акалькулия. Постепенно развивается глубокая социальная дезадаптация. Отличительной особенностью лобно-височной деменции является относительная сохранность памяти и пространственной ориентации.
Двигательные нарушения у больных с лобно-височной деменцией манифестируют в среднем через 4—8 лет после появления первых симптомов болезни. Наиболее типично развитие синдрома паркинсонизма, проявляющегося брадикинезией, гипомимией, гипофонией, мышечной ригидностью. Тремор покоя отсутствует. В некоторых случаях быстро прогрессирующий паркинсонизм может быть ведущим клиническим синдромом с самого начала болезни. Расстройства походки у больных лобно-височной деменцией носят сложный характер в виду сочетания паркинсонизма с апраксией ходьбы (шаркающая, медленная походка на широко расставленных ногах, неустойчивость туловища, затруднения в инициации движения).
У части больных могут наблюдаться пирамидные симптомы, рефлексы орального автоматизма, ами-отрофии, фасцикуляции, парезы глазодвигательной мускулатуры, эпилептические припадки, недержание мочи.
Из дополнительных методов обследования наиболее информативны КТ и МРТ головного мозга, выявляющие атрофические изменения лобно-теменно-височной области и расширение боковых желудочков, особенно передних рогов. При SPECT и ПЭТ обнаруживается снижение кровотока и метаболизма в передних отделах больших полушарий мозга и в области базальных ганглиев.
Лечение лобно-височной деменции не разработано. Препараты леводопы, как правило, неэффективны. Продолжительность болезни не превышает 10-15 лет.
- Читать далее "Паллидо-понто-нигральная дегенерация. Кальцификация базальных ганглиев - болезнь Фара."
Оглавление темы "Наследственные формы атаксии.":1. Дифференцировка болезни телец Леви. Дифференциальная диагностика болезни телец Леви.
2. Болезнь Альцгеймера и диффузных телец Леви. Общие признаки болезни Альцгеймера и диффузных телец Леви.
3. Прогрессирующий паралич и болезнь телец Леви. Болезнь Крейтцфельдта-Якоба и диффузных телец Леви.
4. Лечение болезни телец Леви. Лечение паркинсонизма при болезни телец Леви.
5. Болезнь Мачадо—Джозеф. Азорская болезнь.
6. Дегенерация Галлервордена—Шпатца. Болезнь Галлервордена—Шпатца.
7. Лобно-височная деменция. Диагностика лобно-височной деменции.
8. Паллидо-понто-нигральная дегенерация. Кальцификация базальных ганглиев - болезнь Фара.
9. Х-сцепленная дегенерация - синдром Lubag. Комплекс БАС-Паркинсонизм-Деменция.
10. Тремор. Виды тремора. Физиологический тремор.