
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дегенерация Галлервордена—Шпатца. Болезнь Галлервордена—Шпатца.
Наследственная паллидарная дегенерация Галлервордена—Шпатца (болезнь Галлервордена—Шпатца) — редкое наследственное дегенеративное заболевание нервной системы, впервые описанное в 1922 г. К настоящему времени в литературе имеется около 50 детальных описаний семейных случаев болезни Галлервордена—Шпатца (БГШ), опубликован также ряд наблюдений спорадических случаев заболевания.
Болезнь Галлервордена—Шпатца наследуется по аутосомно-рецессивному типу. Ген, ответственный за возникновение болезни, локализуется на коротком плече 20-й хромосомы в локусе 20р12.3-р13. Первичный молекулярный дефект не установлен.
Патологоанатомически болезнь Галлервордена—Шпатца характеризуется двумя основными признаками:
1) отложением железосодержащего пигмента в области бледного шара, ретикулярной части черной субстанции и красных ядер, что придает им характерное желтовато-коричневое окрашивание;
2) наличием в базальных ганглиях, коре больших полушарий, периферических нервах особых сфероидных нейроаксональных образований, представляющих собой локальные расширения аксонов с пролиферацией мембранных и тубулярных структур.
К неспецифическим изменениям при болезни Галлервордена—Шпатца относятся гибель нейронов в различных отделах полушарий мозга и мозжечка, глиоз, диффузная демиелинизация.
В соответствии с возрастом начала болезни выделяют инфантильные (начало в 1—3 года), типичные детские (4—15 лет) и поздние (старше 16—20 лет) случаи БГШ. Характерный симптомокомплекс БГШ включает прогрессирующие экстрапирамидные, пирамидные и когнитивные нарушения. Нередко могут наблюдаться также пигментная дегенерация сетчатки и атрофия зрительных нервов, стволовые симптомы, амиотрофии, расстройства координации, эпилептические припадки.
При ранней форме болезни Галлервордена—Шпатца заболевание обычно начинается с изменений походки, повышения тонуса в ногах, с последующим развитием генерализованного паркинсоноподобного синдрома и (или) тяжелых дистонических гиперкинезов. Характерны дизартрия и дисфония, обусловленные поражением стволовых структур, оромандибулярная дистония. Могут наблюдаться также миоклония, хореиформный гиперкинез, тремор.
Психические расстройства, отмечаемые уже на ранней стадии болезни, проявляются снижением успеваемости детей в школе, агрессивностью, асоциальным поведением, нарушением памяти, снижением круга интересов. Именно для детской формы БГШ наиболее типичны указанные выше расстройства зрения, эпилептические припадки, поражения черепных нервов.
Совершенно по-другому может протекать поздняя форма болезни Галлервордена—Шпатца, особенно при манифестации симптомов в конце второго десятилетия жизни или после 20 лет. В этих случаях в клинической картине преобладает синдром паркинсонизма, характеризующийся гипокинезией, ригидностью, тремором покоя, постуральной неустойчивостью. Отличительной особенностью паркинсонизма при позднем варианте БГШ является сочетание с дистонией, пирамидными симптомами, прогрессирующей деменцией.
В то же время расстройства зрения, эпилептические припадки, амиотрофии у этих больных встречаются реже, чем при ранней форме заболевания.
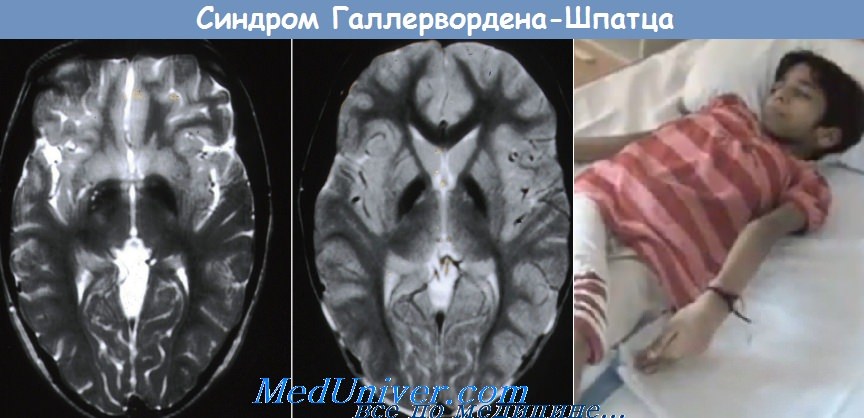
Важнейшее значение в прижизненной диагностике болезни Галлервордена—Шпатца имеет МРТ головного мозга, выявляющая симметричное снижение интенсивности сигнала в Т2-режиме в области бледного шара и ретикулярной части черной субстанции, обусловленное накоплением железа, нередко с дополнительным наличием небольшой зоны гиперинтенсивного сигнала в передне-медиальной части бледного шара (следствие гибели нейронов, глиоза и демиелинизации). Это придает MP-картине на аксиальных срезах в области бледного шара характерный вид «глаза тигра».
Другие изменения вещества мозга менее специфичны и соответствуют картине диффузного атрофического процесса, вовлекающего большие полушария, ствол мозга, мозжечок. При КТ иногда дополнительно выявляются одно- или двусторонние кальцификаты во внутреннем сегменте бледного шара. У части больных, особенно в начале болезни, изменения при КТ и МРТ могут отсутствовать.
Заболевание характеризуется неуклонно прогрессирующим течением, однако темп прогрессирования и длительность болезни существенно различаются при разных клинических формах. Чем раньше дебют заболевания, тем тяжелее его течение и хуже прогноз. При детских формах продолжительность заболевания составляет 6—15 лет. При более позднем развитии болезни, особенно при преобладании в клинике «чистого» паркинсонизма и отсутствии выраженной деменции возможно значительно более длительное течение (до 20 и более лет).
В последнем случае назначение препаратов леводопы иногда позволяет на протяжении ряда лет контролировать выраженность гипокинезии, уровень физической активности больных и повышает возможности самообслуживания. Дополнительно назначают антиконвульсанты (при эпилептических припадках), холинолитики (при дистонии).
- Читать далее "Лобно-височная деменция. Диагностика лобно-височной деменции."
Оглавление темы "Наследственные формы атаксии.":1. Дифференцировка болезни телец Леви. Дифференциальная диагностика болезни телец Леви.
2. Болезнь Альцгеймера и диффузных телец Леви. Общие признаки болезни Альцгеймера и диффузных телец Леви.
3. Прогрессирующий паралич и болезнь телец Леви. Болезнь Крейтцфельдта-Якоба и диффузных телец Леви.
4. Лечение болезни телец Леви. Лечение паркинсонизма при болезни телец Леви.
5. Болезнь Мачадо—Джозеф. Азорская болезнь.
6. Дегенерация Галлервордена—Шпатца. Болезнь Галлервордена—Шпатца.
7. Лобно-височная деменция. Диагностика лобно-височной деменции.
8. Паллидо-понто-нигральная дегенерация. Кальцификация базальных ганглиев - болезнь Фара.
9. Х-сцепленная дегенерация - синдром Lubag. Комплекс БАС-Паркинсонизм-Деменция.
10. Тремор. Виды тремора. Физиологический тремор.