
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
1. Болезнь Гоше (глюкоцеребральный липидоз, острая неврологическая болезнь Гоше, болезнь Гоше, тип II). Манифестация вскоре после рождения: выраженная гипотрофия, бульбарные расстройства, беззвучный крик, нарушения глотания, расстройство глазодвигательных нервов, быстро прогрессирующее снижение зрения, пигментная дегенерация сетчатой оболочки глаза, нарастающая спастическая ригидность, децеребрация.
Смерть наступает к 1-2 году жизни. Патоморфологически: гепатоспленомегалия, в ткани печени обнаруживаются клетки Гоше, накопление в тканях внутренних органов глюкоцереброзидов. В основе заболевания - (3-галактозидазная недостаточность. Тип наследования — А/Р. Дифференциальный диагноз проводится с другими формами нейролипидозов. Важное диагностическое значение приобретают такие симптомы, как отсутствие «вишневой косточки» на глазном дне, скелетных аномалий, лицевого дисморфизма и вакуолизированных лимфоцитов.
Дифференциальный диагноз проводится с Gm2-ганглиозидозом I типа, хронической формой болезни Гоше I типа, инфантильной формой болезни Нимана-Пика.
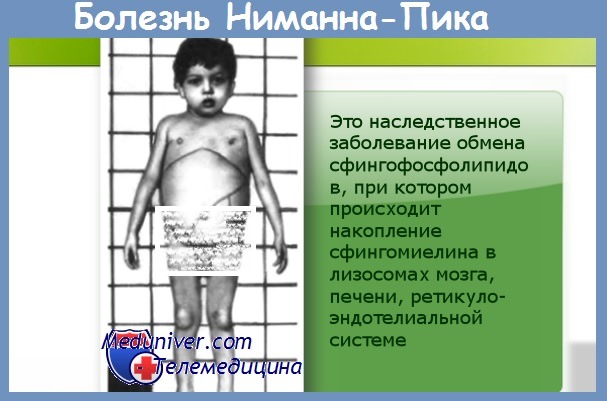
2. Ранняя инфантильная форма болезни Нимана-Пика (сфингомиелиновый липидоз, болезнь Нимана-Пика, тип А). Манифестация вскоре после рождения: анорексия, гипотрофия, анемия, часто бронхопневмония.
Постепенно снижаются слух, зрение, интеллект, появляются спастический тетрапарез, атрофия зрительных нервов, «вишневая косточка» на глазном дне, снижение зрения или слепота. Смерть наступает на 1-4-м году жизни. Патоморфологически: гепатоспленомегалия, накопление сфингомиелина в тканях, обнаруживаются пенистые клетки. В основе заболевания - недостаточность сфингомиелиназы.
Тип наследования — А/Р. Дифференциальный диагноз проводится с болезнью Тея-Сакса, генерализованным ганглиозидозом (Gm1-ганглиозидоз, тип), инфантильной формой болезни Гоше, циррозом печени и лимфагранулематозом.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера"
Оглавление темы "Наследственные метаболические энцефалопатии у грудных детей":- Биохимическиая характеристика наследственных аминоацидопатий
- Метаболические энцефалопатии грудных детей: болезни Тея-Сакса и Сандхоффа
- Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
- Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
- Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса
- Метаболические энцефалопатии грудных детей: болезни Лея, Менкеса, врожденный лактатацидоз
- Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
- Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы
- Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
- Дифференциация метаболических энцефалопатий грудных детей