
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
1. Болезнь Целльвегера (цереброгепаторенальный синдром). Манифестация вскоре после рождения. У детей, имеющих нормальную массу тела, определяются мышечная гипотония, затрудненное сосание и глотание. Отмечается задержка психомоторного развития; церебральная дисфункция сочетается с множественными аномалиями скелета, глаз, половых органов и зрения.
Нередко выявляются контрактуры, деформации суставов, глаукома, катаракта, гепатомегалия. Смерть наступает в раннем возрасте. Патоморфологически обнаруживают множественные аномалии головного мозга, кисты. Патогенез заболевания неизвестен. В ряде случаев находят значительное снижение N-ацетиласпартата. Тип наследования — А/Р. Дифференциальный диагноз проводится с врожденными хондродистрофиями и нарушениями процесса кальцификации.
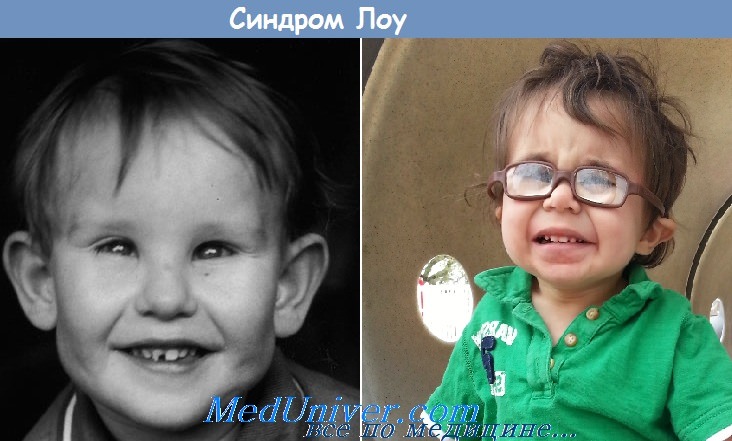
2. Синдром Лоу (окулоцереброренальный синдром). Манифестация в первые месяцы жизни: задержка психомоторного развития, мышечная гипотония, явления дегидратации, нарушение окостенения скелета, остеопороз. С рождения отмечаются глазные симптомы: катаракта, глаукома, хориоретинальные очаги, нистагм, слепота.
В моче обнаруживаются выраженная аминоацидурия, глюкозурия, протеинурия, высокая концентрация пировинограднои кислоты, хлоридов. Смерть наступает в раннем возрасте. Патоморфологически находят аномалии строения коры и сосудов головного мозга, деструкцию тубулярного эпителия почек и грубые изменения глаз. Специфический дефект не установлен; считают, что в основе заболевания лежит нарушение канальцевой реабсорбции. Тип наследования — рецессивный, сцепленный с Х-хромосомой. Дифференциальный диагноз проводится с другими врожденными синдромами.
3. Гликогеноз I типа (нефромегальный гликогеноз, болезнь Гирке). Манифестация вскоре после рождения: анорексия, рвота, гипогликемия, судороги, респираторный дистресс-синдром, гепатомегалия, нефромегалия, стеаторрея, кетонурия, склонность к инфекционным заболеваниям, задержка психомоторного развития.
В основе заболевания - отсутствие глюкозо-6-фосфатазы в печени и почках, накапливание нерасщепленного гликогена в тканях. Смерть наступает в раннем возрасте. Тип наследования — А/Р.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы"
Оглавление темы "Наследственные метаболические энцефалопатии у грудных детей":- Биохимическиая характеристика наследственных аминоацидопатий
- Метаболические энцефалопатии грудных детей: болезни Тея-Сакса и Сандхоффа
- Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
- Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
- Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса
- Метаболические энцефалопатии грудных детей: болезни Лея, Менкеса, врожденный лактатацидоз
- Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
- Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы
- Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
- Дифференциация метаболических энцефалопатий грудных детей