
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
Исследования последних лет дополнили эту группу нозологических форм, которые по срокам манифестации и тяжести поражений очень близки к хорошо известным. К ним относятся гликогеноз \/ типа и недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи.
Гликогеноз V типа (гликогеноз мышечный, болезнь Мак-Ардога) обусловлен врожденным дефицитом миофосфорилазы. Ген для миофосфорилазы (PGYM) относится к хромосоме 11 и имеет 33 различных мутации. Мутации обнаруживаются в различных этнических группах. Заболевание манифестируете детском возрасте, течение прогрессирующее, ведущее к образованию мышечных контрактур. Клинически проявляется нарастающей мышечной слабостью, повышенной утомляемостью. У большинства больных отмечается миоглобинурия. Наследуется по аутосомно-рецессивному типу. В лечении используется диета, богатая белком, и аэробика, возлагаются надежды на генную терапию.
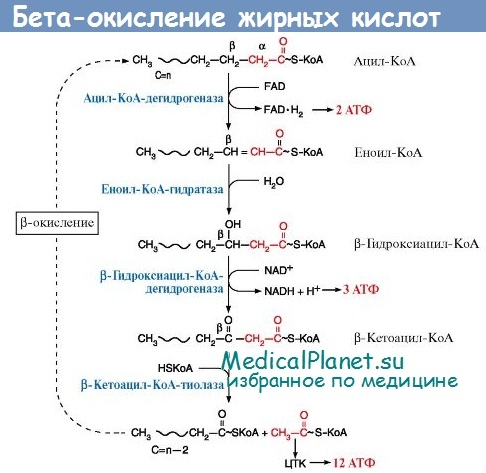
Недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи. Относится к группе болезней клеточной биоэнергетики, состоящей из 10 отдельных нозологических форм, обусловленных дефектами окисления жирных кислот. Частота 1:6400 новорожденных. Тип наследования — аутосомно-рецессивный, ген картирован на коротком плече хромосомы 1 влокусе 1 р 31. Мутация отличается гетерогенностью, существует более 20 вариантов мутан-тных аллелей, один из которых оценивается как второй по частоте в европейской популяции (1:40 или 1:60).
Манифестация наступает в возрасте 6 мес - 2-х лет жизни. Провоцируется, как правило, интеркуррентными заболеваниями и голодом. Начало болезни носит острый характер: повторная рвота, учащенный стул, генерализованные тонико-клонические судороги, прогрессирующая вялость, сонливость, переходящая в кому. Биохимические нарушения сводятся к грубой гипогликемии, умеренному ацидозу, повышенной активности трансаминаз, уровней аммиака и молочной кислоты крови, низкому содержанию карнитина, умеренной кетонурии.
Диагностика основана на выявлении характерного спектра органических кислот мочи, определении низкой активности ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи в лейкоцитах или фибробластах, а также в выявлении мутантныхаллелей методом полимеразной цепной реакции.
Основным методом профилактики приступов болезни являются избегание голода, правильное питание и обогащение пищевого рациона углеводами и прием препаратов карнитина.
- Рекомендуем далее ознакомиться со статьей "Дифференциация метаболических энцефалопатий грудных детей"
Оглавление темы "Наследственные метаболические энцефалопатии у грудных детей":- Биохимическиая характеристика наследственных аминоацидопатий
- Метаболические энцефалопатии грудных детей: болезни Тея-Сакса и Сандхоффа
- Метаболические энцефалопатии грудных детей: болезни Гоше, Нимана-Пика
- Метаболические энцефалопатии грудных детей: лейкодистрофии Краббе, Пилицеуса-Мерцбахера, болезнь Фарбера
- Метаболические энцефалопатии грудных детей: болезни Канаван-ВанБогорта-Бертрана, Александера, Альперса
- Метаболические энцефалопатии грудных детей: болезни Лея, Менкеса, врожденный лактатацидоз
- Метаболические энцефалопатии грудных детей: болезни Целльвегера, Гирке, синдром Лоу
- Метаболические энцефалопатии грудных детей: болезнь Помпе, дефицит гликоген-синтетазы, непереносимость фруктозы
- Метаболические энцефалопатии грудных детей: болезнь Мак-Ардога, недостаточность ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи
- Дифференциация метаболических энцефалопатий грудных детей