
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Наследственная копропорфирия (НКП) - этиология, клиника, диагностика, лечение
Наследственная копропорфирия - краткий обзор:
- Очень редкое заболевание (зарегистрировано менее 50 случаев).
- Аутосомно-доминантное заболевание, вызванное дефицитом копропорфириноген-оксидазы.
- Начало на втором или третьем десятилетии жизни; редко возникает до пубертатного периода.
- Кожные симптомы, неотличимые от таковых при поздней порфирии кожи, встречаются менее чем у 10% пациентов; острые приступы, сходные с таковыми при острой интермиттирующей порфирии являются наиболее типичным признаком (нейро-дермальная порфирия).

а) Эпидемиология. Хотя наследственная копропорфирия (НКП) является редким заболеванием, оно встречается во всем мире, как и острая интермиттирующая порфирия (ОИП), и чаще поражает женщин, чем мужчин. На настоящее время зарегистрировано менее 50 пациентов. Помимо классической наследственной копропорфирии, была описана крайне редко встречающая форма порфирии, названная хардеропорфирия. Это заболевание является гомозиготным вариантом наследственной копропорфирии (НКП), при котором активность копроген-оксидазы очень низка (10% от контрольных значений).
б) Этиология. Наследственная копропорфирия (НКП) — аутосомно-доминантное заболевание, причиной которого является снижение остаточной каталитической активности копроген-оксидазы. Фермент располагается в межмембранном пространстве митохондрии, имеет молекулярную массу 74 кДа и катализирует превращение копрогена III в протоген. Копроген III имеет четыре карбоксильные группы, каждая из которых представляет собой часть пропионатной боковой цепи.
Копроген-оксидаза катализирует последовательное окислительное удаление карбоксильной группы от двух из пропионатных групп, образуя первый 3-карбоксильный порфириноген, известный как хардеропорфириноген (хардероген) и следующий 2-карбоксипорфириноген, известный как протоген.
Копрогеноксидаза кодируется структурным геном с аналогичным названием — геном копроген-оксидазы (СРОХ), как предполагается саузерн-блоттингом ограниченных фрагментов человека. СРОХ находится на хромосоме 3q12, занимает геномное пространство 14 кб и состоит из семи экзонов и шести интронов. У пациентов с гетеро- и гомозиготным вариантами наследственной копропорфирии были обнаружены разнообразные мутации.
К ним относятся точечные мутации, приводящие к основным заменам или пропускам экзонов. Это приводит к повышению экскреции копропорфирина с мочой и калом, что предполагает наличие единого гена.
в) Клиника наследственной копропорфирии (НКП). Как и при пестрой порфирии (ПП), спектр клинических симптомов включает в себя как кожные проявления, так и жизнеугрожающие острые неврологические приступы. Кожные симптомы возникают менее чем у 10% пациентов и являются клинически неотличимыми от таковых при поздней порфирии кожи (ППК) и пестрой порфирии.
Острые приступы идентичны таковым при острой интермиттирующей порфирии (ОИП), и при наследственной копропорфирии преобладают нейровисцеральные симптомы: боль в животе, рвота, запоры, нейропатии и психиатрические реакции. Пероральный гипогликемический агент глипизид, как было указано, отягощает копропорфириеподобный синдром, обратимый после прекращения приема препарата.
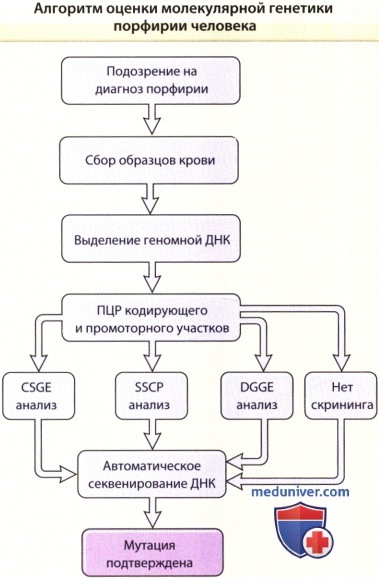
CSGE — конформационно-чув-ствительный гель-электрофорез; DGGE — денатурирующий градиентный гелевый электрофорез;
ПЦР — полимеразная цепная реакция; SSCP — полиморфизм конформации одноцепочечной ДНК.
г) Анализы при наследственной копропорфирии (НКП). ВКП характеризуется повышенной экскрецией КОПРО III с мочой и калом. У таких пациентов уровень КОПРО в кале, более 90% которого представлено изомером III, постоянно значительно повышен (обычно в 10-100 раз). К тому же, фекалии могут также содержать увеличенное количество гепта-, гекса- и пентакарбоксильных порфиринов. Уровень КОПРО III в моче также повышается во время приступов, как и уровни предшественников АЛК и ПБГ. Последние могут быть в нормальных пределах в межприступном периоде.
д) Гистология. Гистологические изменения отсутствуют.
е) Дифференциальная диагностика. При наследственной копропорфирии кожные проявления встречаются редко и даже если и возникают, то у гомозиготных пациентов с манифестом в детском возрасте; в таком случае они напоминают симптоматику при ППК и пестрой порфирии. Преобладание в кале КОПРО III скорее говорит о НКП, чем о ПП, при котором обычно повышены уровни ПРОТО и КОПРО.
Острые приступы такие же, как при ОИП. Дифференциальная диагностика основана на определении уровня порфиринов в кале и копроген-оксидазы в фибробластах или лейкоцитах. Также может проводиться анализ ДНК на наличие мутаций в гене копроген-оксидазы.
ж) Лечение наследственной копропорфирии (НКП). Лечение такое же, как при ОИП, и ПП и описано в таблице ниже.

- Рекомендуем далее ознакомиться со статьей "Порфирия, связанная с дефицитом дегидратазы δ-аминолевулиновой кислоты (АЛК-дегидратазы, ALAD)"
Редактор: Искандер Милевски. Дата публикации: 2.11.2018