
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Врожденная эритропоэтическая порфирия (ВЭП) - этиология, клиника, диагностика, лечение
Врожденная эритропоэтическая порфирия (ВЭП) - краткий обзор:
- Очень редкое заболевание (зарегистрировано около 200 пациентов).
- Аутосомно-рецессивное заболевание, вовлекающее дефицит уропорфириноген III синтазы.
- Начало заболевания в грудном возрасте или на первом десятилетии жизни.
- К кожным симптомам относятся везикулы и буллы, эрозии, корочки, милиа, обезображивающее рубцевание, гиперпигментация и гипертрихоз.
- К системным признакам относятся гемолитическая анемия и гепатоспленомегалия.
- Отложения порфирина встречаются в костях и зубах (эритродонтоз).

а) Эпидемиология. Врожденная эритропоэтическая порфирия (ВЭП) встречается во всем мире. Зарегистрировано менее 500 случаев. Практически всегда болезнь начинается в детстве, однако манифестация во взрослом возрасте описана в 13 случаях. Врожденная эритропоэтическая порфирия (ВЭП) часто наблюдается у животных, в том числе у свиней, крупного рогатого скота и кошек, и исследования на моделях этих животных значительно помогло в изучении этого заболевания.
б) Этиология. Врожденная эритропоэтическая порфирия (ВЭП) — заболевание с аутосомно-рецессивным типом наследования, которое развивается вследствие дефицита различной степени уроген-III-синтазы — четвертого фермента в процессе синтеза гема, катализирующего образование урогена III из гидроксиметилбилана или ПБГ в присутствии ПБГ-деаминазы. Единственным различием меду изомерами порфириногена I и III является поворот боковых цепей D-кольца этого тетрапиррола.
Простой инверсией одной молекулы ПБГ во время синтеза урогена I образуется изомер III. Эта очевидно меньшая структурная вариация имеет огромное биологическое значение, так как лишь изомеры III могут образовывать гем. Образование урогена III катализируется цитоплазматическим ферментом уроген-III-синтазой, тесно связанной с ПБГ-деаминазой. Этот фермент был очищен от эритроцитов человека, является мономером и имеет молекулярную массу почти 29,5 кДа; он термолабилен и обладает максимальной каталитической активностью при pH 7,4.
В большинстве тканей уроген-III-синтаза присутствует в значительном избытке по сравнению с ПБГ-деаминазой, что обеспечивает эффективное превращение в изомер III. Печеночная и эритроцитарная формы фермента идентичны.
Человеческая уропорфириногенсинтаза (UROS) — это цитозольный белок, закодированный единственной копией одноименного гена, обозначаемого UROS, который состоит из 10 экзонов и расположен на хромосоме 10q25.2-q26.3. Человеческая кДНК уроген-III-синтазы была клонирована и секвенирована, она кодирует белок, состоящий из 265 аминокислот. На настоящее время, было выделено почти 30 мутаций в генеуроген-III-синтазы (UROS), включая несколько в эритроидно-специфическом промоторе.
Недостаточность уропорфиририногенсинтазы проявляется накоплением гидроксиметилбилана, спонтанно переходящего в уропорфирин-1. Наследственная эритропоэтическая порфирия также встречается в сочетании с талассемией вследствие новой мутации зародышевой линии в Х-сцепленного эритроидспецифического фактора транскрипции GATA-связывающего белка I типа (GATA-1).
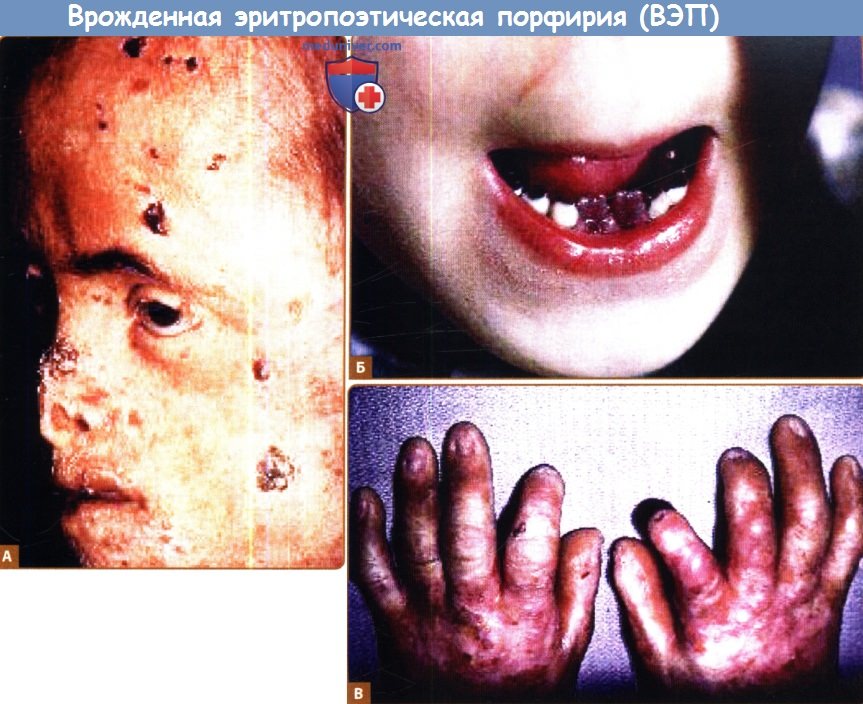
А, Б. Выраженное рубцевание носовых хрящей, потеря волос и гипопигментация зубов (эритродонтия).
В. Хрупкость кожи, образование пузырей, эррозий и контрактур вследствие тяжелого рубцевания в области рук.
в) Клиника врожденной эритропоэтической порфирии (ВЭП). Заболевание обычно развивается в течение первых нескольких месяцев жизни, с умеренной или тяжелой кожной фоточувствительностью, сочетающейся с розово-красным цветом мочи. У пациентов отмечается повышенный уровень УРО I и КОПРО I в эритроцитах, плазме и коже.
К кожным проявлениям относятся повышенная ранимость кожи, везикулы и буллы, которые могут содержать розовую флуоресцентную жидкость. Часто встречаются вторичное инфицирование, замедленное заживление и рубцевание. Это может привести к потере дистально расположенных тканей, например, кончика уха, носа или пальцев, а также деформациям лица. Может встречаться гирсутизм с длинными, темными, лануго-подобными волосами, что особенно выражено на участках кожи, подверженных солнечному воздействию, таких как лицо, шея и конечности. На волосистой части кожи головы может наблюдаться облысение вследствие заживления поражений.
К другим симптомам, возникающим в течение болезни, относятся изменения со стороны глаз (фотофобия, кератоконъюнктивит, эктропион, симблефарон и даже потеря зрения) и пятнистая гипери гипопигментация кожи. Эритродонтоз (красная окраска зубов) является частым симптомом, поражающим как молочные, так и постоянные зубы, и представляет патогнопоничный для ВЭП признак. Цвет мочи также может стать розовато-красным.
К сочетанным внекожным проявлениям относятся спленомегалия, насыщенный порфирином холелитиаз и флуоресцентные нормобласты костного мозга. Встречаются гемолитическая анемия с укороченной продолжительностью жизни эритроцитов (36 дней против 120 дней в норме) и гиперспленизм. Связана ли гемолитическая анемия с «фотогемолизом» циркулирующих порфирин-содержащих эритроцитов или с сочетанным интракорпускулярным дефектом эритроцитов, остается неустановленным.
г) Лабораторные данные. Первичный дефект при ВЭП заключается в сниженной активности уроген-III-синтазы, что приводит к накоплению в тканях порфиринов преимущественно I типа. Часто можно выявить розовый или даже бордовый цвет мочи вследствие избытка УРО I. Нормобласты костного мозга демонстрируют относительно стабильное свечение и содержат значительно повышенные уровни УРО I, КОПРО I и ПРОТО. Экскреция с мочой АЛК и ПБГ находится в нормальных пределах. КОПРО I может в больших количествах находиться в кале. Типичные биохимические признаки ВЭП обобщены в таблице ниже.
д) Гистология врожденной эритропоэтической порфирии (ВЭП). Буллезный элемент при ВЭП является субэпидермальным с минимальной степенью воспаления. В участках рубцевания может наблюдаться утолщение коллагеновых волокон. При исследовании неокрашенных образцов кожи или печени под флуоресцентным микроскопом могут обнаруживаться периваскулярные депозиты порфирина.
е) Дифференциальная диагностика. Диагноз ВЭП обычно может быть поставлен на основании раннего начала тяжелой кожной фоточувствительности в сочетании с красновато-розовым свечением мочи и эритродонтозом. Важно отличить ВЭП от других врожденных фотодерматозов. Кожные симптомы у пациентов с пигментной ксеродермой, буллезным эпидермолизом, световой оспой и буллезным пемфигоидом могут напоминать таковые при ВЭП, но определение уровня порфирина поможет их дифференцировать.
У пациентов с хроническими печеночными порфи-риями,такими как ППК и ПП, эритроцитарного ПРОТО находятся в нормальных пределах. Кожные признаки ГЭП могут иметь поразительное сходство с таковыми при ВЭП, и чтобы отличить эти два заболевания необходимо определить уровни эритроцитарной уроген-Ш-синтазы (снижена при ВЭП) и уроген-декарбоксилазы (снижена при ГЭП). Был опубликован случай сочетания НКП и ВЭП у одной пациентки, которая унаследовала ген НКП от своей матери, а ВЭП от обоих родителей.

ж) Лечение. Терапия при ВЭП главным образом профилактическая и симптоматическая и включает в себя избегание солнечного света, контроль анемии и лечение повторяющихся кожных инфекций. Абсолютная защита от солнечного света с применением фотопротективной одежды может приводить к существенному успеху. Эффективность топических солнцезащитных средств доказана не была. Пероральный β-каротин неэффективен. Спленэктомия, выполняемая для купирования трудно поддающейся терапии гемолитической анемии, в редких случаях приводила к значительному улучшению как анемии, так и кожной фоточувствительности.
Подавление эритропоэза с помощью трансфузии эритроцитарной массы может уменьшить продукцию и выделение порфирина, однако в таких случаях встречается вторичная перегрузка железом. Пероральный прием активированного угля (60 г три раза в день) в течение девяти месяцев снизил уровни порфирина в плазме и коже у одного пациента и привел к полной ремиссии фоточувствительности в течение периода лечения. Полагают, что уголь нарушает энтеропеченочную рециркуляцию порфирина. Однако этот эффективный результат был оспорен. Аллогенная трансплантация костного мозга была эффективной у небольшого числа пациентов и, таким образом, по-видимому, является в настоящее время методом лечения первой линии.
- Рекомендуем далее ознакомиться со статьей "Острая интермиттирующая порфирия (ОИП) - этиология, клиника, диагностика, лечение"
Редактор: Искандер Милевски. Дата публикации: 2.11.2018